 2017, Vol. 35
2017, Vol. 35Institute of Oceanology, Chinese Academy of Sciences
Article Information
- HUANG Ronglian(黄荣莲), LI Li(李莉), ZHANG Guofan(张国范)
- Structure-based function prediction of the expanding mollusk tyrosinase family
- Chinese Journal of Oceanology and Limnology, 35(6): 1454-1464
- http://dx.doi.org/10.1007/s00343-017-6066-9
Article History
- Received Mar. 11, 2016
- accepted in principle Jul. 25, 2016
- accepted for publication Sep. 6, 2016




2 University of Chinese Academy of Sciences, Beijing 100049, China;
3 Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266235, China;
4 Marine Pearl Culture Lab, Fishery College, GuangDong Ocean University, Zhanjiang 524088, China
Tyrosinase (Ty; EC 1.14.18.1; monophenol/ diphenol oxygen oxidoreductase) is a metalloenzyme that belongs to the type-3 copper protein superfamily. Ty is distributed widely in various organisms, such as mammals and bacteria for pigmentation (True, 2003; Slominski et al., 2004), in arthropods for host defense (Gillespie et al., 1997), wound healing (Lai-Fook, 1966) and cuticle sclerotization (Andersen, 2010), and in mollusks for host defense (Zhou et al., 2012), periostracum sclerotization (Zhang et al., 2006) and calcification (Nagai et al., 2007).
According to a previous study (Aguilera et al., 2013), the type-3 copper protein superfamily can be divided into three subclasses based on the domain architecture and the conserved residues that constitute the copper-binding sites: (1) secreted (α, subclass), (2) cytosolic (β subclass) and (3) membrane-bound (γ subclass) subclasses. All three types are considered to have evolved from an ancestral secreted Ty. Subtle changes of the amino acid composition of the binuclear copper active site led to differing functionalities in extant hemocyanins (Hcs), Tys (cytosolic and membrane-bound) and catechol oxidases (CaOs). In arthropods prophenoloxidases (PPOs), which are inactive states of Tys and CaOs, and Hcs are not easily distinguished (Decker and Tuczek, 2000). Decker and Tuczek (2000) used Hcs as model systems to understand substrate-active site interactions between CaOs and Tys by structural analysis (Decker and Tuczek, 2000). The first crystal structure of Ty from Streptomyces castaneoglobisporus was determined and provided information supporting functional differences among members of the type-3 copper-binding protein family (Decker et al., 2006; Matoba et al., 2006). Recently, functional diversity of the molluscan Ty has received increasing interest. Comparative genomics of two bivalve lineages (Crassostrea gigas and Pinctada spp.) have revealed that the Ty gene family is substantially larger than originally thought (Zhang et al., 2012; Aguilera et al., 2014). The cause of intraspecies and interspecies functional diversities among these enzymes has also been debated. For example, Cu ligand exchange is a major factor of Ty functional diversity in mammals (Aguilera et al., 2014). However, the absence of information detailing the structure of Tys remains a major limitation for investigating the diverse functions of these enzymes. Furthermore, omics data have increased, requiring an efficient method to accelerate Ty structure-function relationship research.
High-throughput structural genomic initiatives that include computational methods are now widely used to predict protein structures of a target sequence and through these predictions a greater understanding of the biological function of a target protein can be obtained (Baker and Sali, 2001). Pho reported single nucleotide polymorphisms (SNPs) or genetic variants in at least nine pigmentation genes including TYR and TYRP1, which are associated with risk for melanoma, other cutaneous malignancies and photosensitivity, by Genome-wide association studies (GWAS) (Pho and Leachman, 2010). The Kamaraj research team has also provided a wealth of valuable information about genetic mutations of TYR (Kamaraj and Purohit, 2013a), TYRP1 (Kamaraj and Purohit, 2013b), OCA2 (Kamaraj and Purohit, 2014a), SLC24A5 (Kamaraj and Purohit, 2014b) and SLC45A2 (Kamaraj and Purohit, 2016) in mammals by in silico screening and molecular dynamics simulations. These examples highlight how computational efforts have provided efficient tools for analysis and evaluation of genetic mutations in proteins that give rise to functional changes with pathological consequences and for revealing their underlying molecular mechanism. The development and application of structure-based function prediction methods have also been discussed (Gherardini and Helmer-Citterich, 2008). Phyre2 (Kelley et al., 2015), i-TASSER (Roy et al., 2010), Swiss-Model (Arnold et al., 2006), HHpred (Söding, 2005), PSI-BLASTbased secondary structure prediction (PSIPRED) (Lobley et al., 2009), Robetta (Raman et al., 2009) and Raptor (Källberg et al., 2012) are the most commonly used web servers to model proteins.
In this study, 12 members of Ty were verified and characterized in C. gigas and Pinctada fucata martensii to obtain additional functional data that describes the expanding Ty family among bivalves. Three-dimensional (3D) structures of Tys were constructed by template-based homology modeling, and based on these models potential functional characteristics and residues involved in enzyme catalysis were identified. Furthermore, the relationships between these characteristics and active sites were analyzed by an online web server that offers a structure-based functional interpretation. Although results of the phylogenetic analysis and expression pattern indicate that Ty genes undergo neofunctionalization in shell formation (Aguilera et al., 2014), structure-related supporting data is still needed.
2 MATERIAL AND METHOD 2.1 AnimalsThe Pacific oysters used for cloning and RT-qPCR were 2 years old and 9–12 cm in shell length, and were purchased from a Qingdao oyster farm. Adult Pearl oysters (~2 years of age and 6–8 cm in shell length) were obtained from Liushagang, Zhanjiang, Guangdong, China. Oysters were cultured in an aquarium at the Institute of Oceanology, Chinese Academy of Sciences (IOCAS). We complied with institutional ethical use protocols for aquatic species (Mason and Matthews, 2012).
2.2 cDNA cloningcDNAs of Tys were cloned, as described previously (Shen, 1997; Yan et al., 2014). Total RNA was prepared using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. RNA quantity was evaluated by measuring OD260/OD280 with a NanoDrop ND1000 spectrophotometer (Thermo scientific, Waltham, MA, USA), while RNA integrity was determined by fractionation on a 1.0% agarose gel and staining with ethidium bromide. The sequences of the Ty genes were obtained from genome of C. gigas (Zhang et al., 2012) and the transcriptome of P. fucata martensii (Zhao et al., 2012). Specific primers were designed based on these fragments. Primer sequences are available upon request. To increase specificity and sensitivity, nested-PCR was performed.
The single-strand cDNA for all RACE reactions was prepared from total RNA of the mantle using the SMART RACE cDNA amplification kit (TaKaRa, Dalian, China), according to the manufacturer's instructions. For the first PCR, the synthesized cDNA was used as the template. For the second PCR, the first amplified product was used as the template. The PCR program was: 94℃ for 5 min, 30 cycles of 94℃ for 30 s, 94℃ for 30 s, 68℃ for 30 s and 72℃ for 1 min, and a final elongation step of 72℃ for 10 min.
2.3 Phylogenetic analysisThe deduced proteins from the identified sequences of Tys in oyster and pearl oyster were aligned by the Clustal algorithm with functionally identified protein sequences of other mollusk species, including P. fucata (synonym of P. fucata martensii), P.margaritifera, P.maxima, Pernaviridis, Azumapecten (Chlamys) farreri, Lottia gigantea, Illex argentinus and Sepia officinalis. The functionally identified Tys are: (1) OT47, likely involved in periostracum formation (AAZ66340.1) (Zhang et al., 2006); (2) Pfty1 and Pfty2, extracted from a prismatic shell layer (BAF42771.1, BAF42772.1) (Nagai et al., 2007); (3) PfTy, highly expressed in the mantle, indicating that this enzyme participates in nacreous layer formation (BAF74507.1) (Takgi and Miyashita, 2014); (4) TYRO1_PINMG (H2A0L0.1), TYRO2_PINMG (H2A0L1.1) and TYRO_PINMA (P86952.1), isolated from a prismatic shell layer (Marie et al., 2012); (5) Tys from the foot of green mussel (AGZ84286.1, AGZ84289.1, AGZ84288.1, AGZ84290.1, AGZ84287.1) (Guerette et al., 2013); (6) AfaTy from the hemolymph of scallop (ACF25906.1) (Zhou et al., 2012); (7) Tys from a shell of limpet (Lotgi1|166196) (Mann et al., 2012); (8) ST94 isolated from the ink of squid, Illex argentinus (BAC87844.1, BAC87843.1) (Naraoka et al., 2003); and (9) Ty from the ink gland of cuttlefish, Sepia officinalis (CAC82191.1). A phylogenetic tree was constructed according to Aguilera et al. (2014) with slight modifications. Neighbor-joining (NJ) reconstructions were performed using MEGA 6.0 with a JTT substitution model and 1 000 bootstrap replicates (Tamura et al., 2013). Maximum-likelihood (ML) trees were also constructed using MEGA 6.0 and the WAG substitution model that started from a random tree and 1 000 bootstrap replicates.
2.4 Molecular modelingConsiderable insight can be gained from structural comparison of a given structure with all other known protein structures. Such analysis can frequently detect structural relationships with functional significance that are not evident from sequence comparisons (Baker and Sali, 2001; Holm and Sander, 1995). Thus, by using the Phyre2 online http://www.sbg.bio.ic.ac.uk/phyre2/ protocol (Kelley and Sternberg, 2009; Kelley et al., 2015), we submitted the conserved binuclear site of Tys to build 3D models. We achieved high-accuracy models with very low sequence identity ( < 30%) by the remote homology/fold recognition of Phyre2. Chimera 1.8.1 was used to display models and PSI-blast pseudomultiple sequence alignment was used for alignment. The Ramachandran plot was used to validate structures based on the C-alpha geometry: phi, psi and C-beta deviation (Hooft et al., 1997; Lovell et al., 2003). The stereochemical qualities of the final models were evaluated by Procheck v3.5 (Laskowski et al., 1993) and Verify-3D (Hooft et al., 1996) with the Swiss Model server (https://swissmodel.expasy.org/docs/structure_assessment).
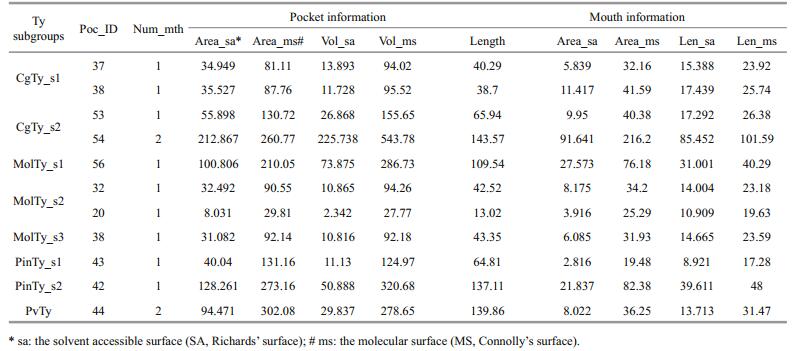
2.5 Active site pocket predictionUsing the CASTp server online http://sts.bioe.uic.edu/castp/ (Dundas et al., 2006), we measured the area and volume of each pocket and cavity of solvent accessible surfaces (SA or Richards' surface) and molecular surfaces (MS or Connolly's surface), number of mouth openings, and area of openings. The results gave information about the location of functionally important residues and facilitated a comprehensive understanding of the structural basis of the protein function. We compared the results with both the mollusk Ty family members and type-3 copper protein superfamily members available in the PDB database.
3 RESULT 3.1 Ty sequencesAlthough a large group of Ty in C. gigas has been defined by a genome project (Zhang et al., 2012), members of Ty family were verified through gene cloning in this study to ensure that the enzymes are functional or are composed of active genes. The 12 mRNAs of Tys composed of 5′ -UTR, 3′-UTR, ORF and deduced primary structure are shown in Fig. 1. Of these mRNAs, 11 contain signal peptides, indicating that these enzymes exhibit secretory characteristics. CgTys have lengths of 373 aa to 742 aa, whereas pma-Ty1 and pma-Ty2 have lengths of 756 and 375 aa, respectively. Based on the sequence alignment of 27 type-3 copper proteins, six histidine residues marked with closed asterisks in CuA and CuB copper-binding domains are conserved in all of the subclasses. The connection between CuA and CuB is a linker region containing two conserved regions, namely, L1 (YWDXXXD) and L2 (GX repeats) (Fig. 1). Phylogenetic analysis showed that the molluscan Ty family is divided into 8 orthologous Ty subgroups (CgTy_s1, CgTy_s2, MolTy_s1, MolTy-s2, MolTy-s3, PinTy-s1, PinTy-s2 and PviTy) (Fig. 1).

|
| Figure 1 Phylogenetic analyses of molluscan Ty family A consensus midpoint-rooted phylogenetic tree based on maximum likelihood (ML) topology is shown. Subgroups are color coded as follows: sienna, CgTy_s1; magenta, CgTy_s2; green, MolTy_s1; dark cyan, MolTy_s2; blue, MolTy_s3; orange, PvTy; yellow, PinTy_s1; red, PinTy_s2. The bold dots with colors indicate the representatives of each subgroup, which were submitted to the Phyre2 server for homologous modeling. NJ bootstrap values (BV) are indicated at the nodes. |
We submitted eight sequences of the two putative copper-binding sites identified in the eight subgroups (marked by colors) to the Phyre server for homology modeling (Fig. 2). Six to Eight templates were selected for homology modeling (Table 1s). Despite low sequence identities (20% to 35%), the final models obtained a 100% confidence match, indicating that the overall fold and the central core of the models are likely to be accurate, even at sequence identities of < 20% (Kelley and Sternberg, 2009). Furthermore, the Ramachandran plot of the final structures showed that 93.6% to 97.9% of all residues in active centers are in favorable regions (Fig. 2s).

|
| Figure 2 Sequence alignment of Ty subgroups and a universal silhouette-rounded ribbon stereomodel of Ty PSI-blast alignment indicate the conserved residues in the eight subgroups (deplayed in asterisk), and the residues involved in the active site pockets and the affiliate pockets (deplayed in green circles and purple boxes) (a); the 1 to 8 cylinders display the helixes in order from N-to C-terminal. Six green His residues cluster in the nuclear of the model (b). |
We constructed a general model of the mollusk Ty using the Chimera software. The model gave the secondary structure and the position of the conserved residues (Figs. 2 and 3). There is the HA1 (8 or 9) HA2XGXXF (3) HA3 motif in CuA and the HB1 (3) HB2 (15) DPXF (3) HB3 motif in CuB. The model contains eight helices (1 to 8) and five loops (not present). In a single globular domain, a four-helix bundle (helices 2, 3, 5 and 8) contains the dicopper active site CuA (HA1, HA3) and CuB (HB1, HB2, HB3), with HA2 missing (Fig. 2b). The two active sites exhibit monophenoloxidase (CuA) or diphenoloxidase activity (CuB). Other conserved residues that are also likely to be functionally important are marked as Z1–Z9. For residues at Z1 and Z7 positions, two phenylalanines are located near the copper site, similar to the F2 and F4 positions in crustacean PPO, which likely stabilizes the copper site conformation and blocks the copper site to prevent non-specific oxidation of phenolic compounds (Masuda et al., 2014). In addition, the conserved sequence pattern Phe-X-X-X-His in all known Hcs constitute the coordination environment of CuA and CuB (Solomon et al., 2014). Z2 (R), Z8 (D) and Z9 (E) clusters in the backbone of helices 3 and 8 possibly act as a "button" that fastens the two-helix backbone by ionic bonding (Fig. 3a), except for CgTy_s2 (Fig. 3b). In Bacillus megaterium Ty (TyBm), this enzyme can absorb a zinc ion in this area (Kanteev et al., 2013). Glu at the Z3 position is highly conserved among mollusk Tys, which are also found in bacterial Ty (E182 in S. castaneoglobisporus) (Matoba et al., 2006) and CaOx (E236 in Catechol oxidases of I. batatas) (Klabunde et al., 1998). Therefore, Glu acts as an important acid/ base residue during catalysis. Z4 is occupied by Asn in seven subgroups of Ty, and replaced with Asp (in A1 and B-Pf subgroups) and Gly (in A3). Z4 is equivalent to Asn 205 of TyBm, which is replaced with either alanine or aspartic acid. Substitutes likely influence the ratio of monophenolase/diphenolase activity (Shuster Ben-Yosef et al., 2010) and significantly reduce the copper uptake rate (Kanteev et al., 2013). The present study revealed the difference at the Z5 position (next to HB2) among mollusk Ty subgroups: Val for PinTy_s2 and MolTy_s3, Asn for B1 and CgTy_s2, Asp for MolTy_s2, and Gln for MolTy_s1. At the same TyBm position, wild-type Arg209 and mutant His209 exhibited different ratios of monophenolase/ diphenolase activity. The mutant contains a more rigid residue that obstructs the active site when compared with that of the wild-type. Thus, the binding of substrate to CuB is inhibited (Sendovski et al., 2011).

|
| Figure 3 Residues variation in CgTy_s2 CgTy_s2 subgroup (in magenta) replaces 3 residues (R, D, E by M, T, I), which are highly conserved in other subgroups (e.g., MolTy_s1 in cyan). Those three residues, R (in Z2 position) and D (Z8), E (Z9) in other subgroups, "button" two helixes (helix 3 and 8) backbone by ionic bonding. |
Two loops likely provide structural stability essential for enzyme activity. In molluscan Hc, the HisA2 ligand of CuA is located on a loop instead of an α-helix and is covalently tethered to a highly conserved Cys residue located at the N-terminus, which is considered necessary to stabilize the His ligand (Cuff et al., 1998; Perbandt et al., 2003). This residue is replaced with various other residues, in particular Ser in mollusk Ty. When a cross-linked Cys mutates, the kcat of L-Ty decreases but does not completely abolish enzymatic function (Fujieda et al., 2011). The other loop is a linker region between helices 6 and 7, which contains a place holder at the Z6 position (L, V, I, Q, A, P, or F) (Fig. 2); as a consequence, the entry of a substrate, such as Val218 in bacterium Ty, is prevented (Sendovski et al., 2011). In contrast to Phe at the Z1 and Z7 positions, the Z6 blocker is not in the vicinity of the active site but on the surface of a dinuclear pocket. This blocker also affects the ratio of monophenolase/diphenolase activity (Goldfeder et al., 2013).
3.3 Locate and measure pockets on protein structuresIn addition to separate analysis of single residues, profiling of binuclear pockets of active sites was performed to visualize the annotated functional residues, with emphasis on mapping the surface pockets and interior voids. Using the CASTp server online, we determined several parameters of the eight subgroups of the mollusk Ty pocket (Table 1), involved residues (Figs. 2 and 4) and electrostatic potential surfaces (Fig. 4). The active site pocket of six subgroups, excluding subgroups MolTy_s3 and MolTy_s1, consists of six His ligands and this active site pocket is coordinated by helices 6 and 7 and the loop between these helices. The most distinct models were found for the CgTy_s1, CgTy_s2 and MolTy_s2 subgroups with an affiliate pocket located in the vicinity of the active site. The affiliate pocket in CgTy types is coordinated by a flexible loop between helices 2 and 3. In contrast, the affiliate pocket for subgroup MolTy_s2 is formed by helices 3 and 5. The common characteristic of the affiliate pockets involves HA2 and Z6 (blocker). Residues at these two positions are also shared by the bicopper active site pocket. The MolTy_s3 type contains the smallest pocket. HA1 in MolTy_s3 is probably concealed by other residues Asn42 and Ile181 (Z6) (black circle in Fig. 4). Moreover, HA3 and HB3 in MolTy_s3 are absent on the surface of the pocket but appear in a closed cavity behind the pocket, which is similar to Hc in Rapana thomasiana (Fig. 5). In the pocket of subgroup MolTy_s1, the absence of a His residue(s) is likely blocked by H47 and F52 (black circle in Fig. 4). According to the measured parameters of those pockets, PvTy and PinTy_s2 contain a relatively large pocket with a multi-opening (Table 1).

|
| Figure 4 An overlay of the electrostatic potential surface and the pocket(s) in active site of Ty subgroups Surface representation colored according to electrostatic potential: red, -12 kT; white, 0 kT; blue, +12 kT. The black circles indicate the blockers in MolTy_s1 (H47 and F52) and MolTy_s3 (N42 and I181) which may affect active core(s) of His residues present on the pocket surface. Those residues involved in pocket forming were emphasized by the amino acid abbreviations. |

|
| Figure 5 The pocket and a closed cavity of Hc in Rapana thomasiana (PDB: 1LNL) (a) and in MolTy_s3 of C. gigas (b) Surface representation colored according to electrostatic potential: red, −12 kT; white, 0 kT; blue, +12 kT. The black asterisk indicates the closed cavity adjacent to the open pocket. |
In previous studies investigating the function of molluscan Ty, gene expression has been frequently used to provide supporting data of diverse functions, such as distinct roles of Tys in C. gigas (Luna-Acosta et al., 2011; Zhang et al., 2012), Pinctada spp. (Felipe Aguilera et al., 2014) and P. viridis (Guerette et al., 2013). However, no high-level structural data, such as crystal structures, have been obtained to reveal detailed functional characteristics at the atomic level. Thus, appropriate structural studies should be conducted to determine structural variations in substrate-binding pockets, substrate accessibility to active sites and enzyme activities. Despite a similar overall structure in binuclear centers, nuclear active pockets vary and different functional residues are used (Figs. 2 and 5). By comparing the active site pocket(s) of the eight Ty subgroups, we found: (1) besides the His residues in the active site, Z1–Z9 represent the most important functional residues in mollusk Tys (Fig. 2); (2) P. viridis byssus types (PvTy), Pinctada spp. shell type (PinTy_s2) and expanded C. gigas shell types (CgTy_s1, CgTy_s2) contain larger or double pockets.
In materials science, shell and byssus in mollusk exhibit excellent material properties, indicating that these materials may share the same molecular designs (Smith et al., 1999). Recent research found Tys that modify tyrosine residues in byssus matrix proteins for self-assembly in P. viridis. Tys in P. viridis participate in adhesive byssus formation by converting tyrosine residues into adhesive DOPA in their substrates, for example PVFP-5, a major plaque protein named Perna viridis foot protein-5 (Suhre et al., 2014). The PSIPRED online prediction tool was used to examine PVFP-5. The results revealed that PVFP-5 is composed of beta sheets and coils (not shown) and Ty-rich fibrous proteins. The Ty residues also maintain DOPA reactivity by providing chemical protection against DOPA oxidation (Yu et al., 2011).
We speculate that for those mantle-specific expression Ty genes, their actual substrate of the C. gigas and P. fucata martensii should also be Ty-rich, fibrous shell proteins, such as CgSP1 (CGI_10007556) and CgSP2 (CGI_10009634) (Fig. 3s). These shell proteins are coil-rich and Ty-rich proteins with the GPY repeat (CgSP1) or YY repeat (CgSP2). Although these proteins contain a low identified peptide number in the shell proteome, coding genes of these proteins are highly expressed in the mantle. This high expression indicates that these proteins are major matrix proteins during shell formation (Fig. 3s). The reason for a low level in the shell is probably limited by enzyme cleavage sites, which are affected by the amino acid sequence and composition. The composition-biased protein lacks enzyme cleavage sites, as predicted by MS-digest (data not shown). This factor subsequently affects the estimation of protein abundance. We also found shell proteins from pearl oyster with a similar feature, including common shell matrix proteins, such as shematrin (Yano et al., 2006), pearlin (Samata et al., 1999), as well as KRMP (McDougall et al., 2013). The shematrin family is composed of major shell matrix proteins in pearl oyster species and these proteins are characterized by a conserved repeat domain designated as XGnX (where X is a hydrophobic amino acid) (Yano et al., 2006). Shematrin members contain GXnY repeats (McDougall et al., 2013) that are similar to those found in other structural proteins, such as spider silks (Hinman and Lewis, 1992). However, mechanisms by which these glycine-rich regions contribute to strength or elasticity of spider silks remain unclear (Breslauer and Kaplan, 2012). In the application area, enzymecatalyzed modification of Bombyx morisilk fibroin, such as Ty (Freddi et al., 2006), horseradish peroxidase and hydrogen peroxide (Partlow et al., 2014), has been investigated to produce a new range of bio-based and environmentally friendly polymers with tunable properties. Despite these efforts, mushroom Ty used in silk fibroin exhibits low activity. Our findings may provide insight into the molecular mechanism of the interaction of Ty and its substrate. The actual functional state of Ty still requires more experimental effort.
5 CONCLUSIONIn conclusion, sequence analysis and structural motif comparison methods support the functional differentiation of the molluscan Ty family. The key residues in the active site of Ty vary between the subgroups. These variations probably give rise to their adaptation to different substrates, such as the shell matrix proteins. Substrate diversity should be considered as an important evolving factor of the molluscan Ty family. Further investigations should focus not only on Tys but also on substrates and their interaction mechanisms.
6 ACKNOWLEDGEMENTWe thank ZHE Zheng for her assistance at the facility.
Electronic supplementary material
Supplementary material (Supplementary Table 1S and Supplementary Figs. 1s–3s) is available in the online version of this article at http://dx.doi.org/10.1007/s00343-017-6066-9.
| Aguilera F, McDougall C, Degnan B M, 2013. Origin, evolution and classification of type-3 copper proteins:lineage-specific gene expansions and losses across the Metazoa. BMC Evolutionary Biology, 13(1): 1–12. Doi: 10.1186/1471-2148-13-1 |
| Aguilera F, McDougall C, Degnan B M, 2014. Evolution of the tyrosinase gene family in bivalve molluscs:independent expansion of the mantle gene repertoire. Acta Biomaterialia, 10(9): 3855–3865. Doi: 10.1016/j.actbio.2014.03.031 |
| Andersen S O, 2010. Insect cuticular sclerotization:a review. Insect Biochemistry and Molecular Biology, 40(3): 166–178. Doi: 10.1016/j.ibmb.2009.10.007 |
| Arnold K, Bordoli L, Kopp J, Schwede T, 2006. The SWISSMODEL workspace:a web-based environment for protein structure homology modelling. Bioinformatics, 22(2): 195–201. Doi: 10.1093/bioinformatics/bti770 |
| Baker D, Sali A, 2001. Protein structure prediction and structural genomics. Science, 294(5540): 93–96. Doi: 10.1126/science.1065659 |
| Breslauer D N, Kaplan D L, 2012. Silks:properties and uses of natural and designed variants. Biopolymers, 97(6): 319–321. Doi: 10.1002/bip.v97.6 |
| Cuff M E, Miller K I, van Holde K E, Hendrickson W A, 1998. Crystal structure of a functional unit from Octopus hemocyanin. Journal of Molecular Biology, 278(4): 855–870. Doi: 10.1006/jmbi.1998.1647 |
| Decker H, Schweikardt T, Tuczek F, 2006. The first crystal structure of tyrosinase:all questions answered. Angewandte Chemie International Edition, 45(28): 4546–4550. Doi: 10.1002/(ISSN)1521-3773 |
| Decker H, Tuczek F, 2000. Tyrosinase/catecholoxidase activity of hemocyanins:structural basis and molecular mechanism. Trends in Biochemical Sciences, 25(8): 392–397. Doi: 10.1016/S0968-0004(00)01602-9 |
| Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J, 2006. CASTp:computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Research, 34(S2): W116–W118. |
| Freddi G, Anghileri A, Sampaio S, Buchert J, Monti P, Taddei P, 2006. Tyrosinase-catalyzed modification of Bombyx mori silk fibroin:grafting of chitosan under heterogeneous reaction conditions. Journal of Biotechnology, 125(2): 281–294. Doi: 10.1016/j.jbiotec.2006.03.003 |
| Fujieda N, Ikeda T, Murata M, Yanagisawa S, Aono S, Ohkubo K, Nagao S, Ogura T, Hirota S, Fukuzumi S, Nakamura Y, Hata Y, Itoh S, 2011. Post-translational His-Cys crosslinkage formation in tyrosinase induced by copper(Ⅱ)-peroxo species. Journal of the American Chemical Society, 133(5): 1180–1183. Doi: 10.1021/ja108280w |
| Gherardini P F, Helmer-Citterich M, 2008. Structure-based function prediction:approaches and applications. Briefings in Functional Genomics and Proteomics, 7(4): 291–302. Doi: 10.1093/bfgp/eln030 |
| Gillespie J P, Kanost M R, Trenczek T, 1997. Biological mediators of insect immunity. Annual Review of Entomology, 42(1): 611–643. Doi: 10.1146/annurev.ento.42.1.611 |
| Goldfeder M, Kanteev M, Adir N, Fishman A, 2013. Influencing the monophenolase/diphenolase activity ratio in tyrosinase. Biochimica et Biophysica Acta, 1834(3): 629–633. Doi: 10.1016/j.bbapap.2012.12.021 |
| Guerette P A, Hoon S, Seow Y, Raida M, Masic A, Wong F T, Ho V H B, Kong K W, Demirel M C, Pena-Francesch A, Amini S, Tay G Z, Ding D W, Miserez A, 2013. Accelerating the design of biomimetic materials by integrating RNA-seq with proteomics and materials science. Nature Biotechnology, 31(10): 908–915. Doi: 10.1038/nbt.2671 |
| Hinman M B, Lewis R V, 1992. Isolation of a clone encoding a second dragline silk fibroin. Nephila clavipes dragline silk is a two-protein fiber. The Journal of Biological Chemistry, 267(27): 19320–19324. |
| Holm L, Sander C, 1995. Dali:a network tool for protein structure comparison. Trends in Biochemical Sciences, 20(11): 478–480. Doi: 10.1016/S0968-0004(00)89105-7 |
| Hooft R W W, Sander C, Vriend G, 1997. Objectively judging the quality of a protein structure from a Ramachandran plot. Computer Applications in the Biosciences, 13(4): 425–430. |
| Hooft R W W, Vriend G, Sander C, Abola E E, 1996. Errors in protein structures. Nature, 381(6580): 272. Doi: 10.1038/381272e0 |
| Källberg M, Wang H P, Wang S, Peng J, Wang Z Y, Lu H, Xu J B, 2012. Template-based protein structure modeling using the RaptorX web server. Nature Protocols, 7(8): 1511–1522. Doi: 10.1038/nprot.2012.085 |
| Kamaraj B, Purohit R, 2013a. Mutational analysis of TYR gene and its structural consequences in OCA1A. Gene, 513(1): 184–195. Doi: 10.1016/j.gene.2012.09.128 |
| Kamaraj B, Purohit R, 2013b. In silico screening and molecular dynamics simulation of disease-associated nsSNP in TYRP1 gene and its structural consequences in OCA3. BioMed Research International, 2013: 697051. |
| Kamaraj B, Purohit R, 2014a. Computational screening of disease-associated mutations in OCA2 gene. Cell Biochemistry and Biophysics, 68(1): 97–109. Doi: 10.1007/s12013-013-9697-2 |
| Kamaraj B, Purohit R, 2014b. Mutational analysis of oculocutaneous albinism:a compact review. BioMed Research International, 2014: 905472. |
| Kamaraj B, Purohit R, 2016. Mutational analysis on membrane associated transporter protein (MATP) and their structural consequences in oculocutaeous albinism type 4 (OCA4)-a molecular dynamics approach. Journal of Cellular Biochemistry, 117(11): 2608–2619. Doi: 10.1002/jcb.v117.11 |
| Kanteev M, Goldfeder M, Chojnacki M, Adir N, Fishman A, 2013. The mechanism of copper uptake by tyrosinase from Bacillus megaterium. JBIC Journal of Biological Inorganic Chemistry, 18(8): 895–903. Doi: 10.1007/s00775-013-1034-0 |
| Kelley L A, Mezulis S, Yates C M, Wass M N, Sternberg M J E, 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols, 10(6): 845–858. Doi: 10.1038/nprot.2015.053 |
| Kelley L A, Sternberg M J, 2009. Protein structure prediction on the Web:a case study using the Phyre server. Nature Protocols, 4(3): 363–371. Doi: 10.1038/nprot.2009.2 |
| Klabunde T, Eicken C, Sacchettini J C, Krebs B, 1998. Crystal structure of a plant catechol oxidase containing a dicopper center. Nature Structural Biology, 5(12): 1084–1090. Doi: 10.1038/4193 |
| Lai-Fook J, 1966. The repair of wounds in the integument of insects. Journal of Insect Physiology, 12(2): 195–226. Doi: 10.1016/0022-1910(66)90136-3 |
| Laskowski R A, MacArthur M W, Moss D S, Thornton J M, 1993. PROCHECK:a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26(2): 283–291. Doi: 10.1107/S0021889892009944 |
| Lobley A, Sadowski M I, Jones D T, 2009. pGenTHREADER and pDomTHREADER:new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics, 25(14): 1761–1767. Doi: 10.1093/bioinformatics/btp302 |
| Lovell S C, Davis I W, Arendall III W B, de Bakker P I W, Word J M, Prisant M G, Richardson J S, Richardson D C, 2003. Structure validation by Cα geometry:?, ψ and Cβ deviation. Proteins, 50(3): 437–450. Doi: 10.1002/prot.10286 |
| Luna-Acosta A, Saulnier D, Pommier M, Haffner P, de Decker S, Renault T, Thomas-Guyon H, 2011. First evidence of a potential antibacterial activity involving a laccase-type enzyme of the phenoloxidase system in Pacific oyster Crassostrea gigas haemocytes. Fish & Shellfish Immunology, 31(6): 795–800. |
| Mann K, Edsinger-Gonzales E, Mann M, 2012. In-depth proteomic analysis of a mollusc shell:acid-soluble and acid-insoluble matrix of the limpet Lottia gigantea. Proteome Science, 10(1): 28. Doi: 10.1186/1477-5956-10-28 |
| Marie B, Joubert C, Tayalé A, Zanella-Cléon I, Belliard C, Piquemal D, Cochennec-Laureau N, Marin F, Gueguen Y, Montagnani C, 2012. Different secretory repertoires control the biomineralization processes of prism and nacre deposition of the pearl oyster shell. Proceedings of the National Academy of Sciences of the United States of America, 109(51): 20986–20991. Doi: 10.1073/pnas.1210552109 |
| Mason T J, Matthews M, 2012. Aquatic environment, housing, and management in the eighth edition of the Guide for the Care and Use of Laboratory Animals:additional considerations and recommendations. Journal of the American Association for Laboratory Animal Science, 51(3): 329–332. |
| Masuda T, Momoji K, Hirata T, Mikami B, 2014. The crystal structure of a crustacean prophenoloxidase provides a clue to understanding the functionality of the type 3 copper proteins. The FEBS Journal, 281(11): 2659–2673. Doi: 10.1111/febs.2014.281.issue-11 |
| Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, Sugiyama M, 2006. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. The Journal of Biological Chemistry, 281(13): 8981–8990. Doi: 10.1074/jbc.M509785200 |
| McDougall C, Aguilera F, Degnan B M, 2013. Rapid evolution of pearl oyster shell matrix proteins with repetitive, lowcomplexity domains. Journal of the Royal Society Interface, 10(82): 20130041. Doi: 10.1098/rsif.2013.0041 |
| Nagai K, Yano M, Morimoto K, Miyamoto H, 2007. Tyrosinase localization in mollusc shells. Comparative Biochemistry and Physiology Part B:Biochemistry and Molecular Biology, 146(2): 207–214. Doi: 10.1016/j.cbpb.2006.10.105 |
| Naraoka T, Uchisawa H, Mori H, Matsue H, Chiba S, Kimura A, 2003. Purification, Characterization and molecular cloning of tyrosinase from the cephalopod mollusk, Illex argentinus. European Journal of Biochemistry, 270(19): 4026–4038. Doi: 10.1046/j.1432-1033.2003.03795.x |
| Partlow B P, Hanna C W, Rnjak-Kovacina J, Moreau J E, Applegate M B, Burke K A, Marelli B, Mitropoulos A N, Omenetto F G, Kaplan D L, 2014. Highly tunable elastomeric silk biomaterials. Advanced Functional Materials, 24(29): 4615–4624. Doi: 10.1002/adfm.201400526 |
| Perbandt M, Guthöhrlein E W, Rypniewski W, Idakieva K, Stoeva S, Voelter W, Genov N, Betzel C, 2003. The structure of a functional unit from the wall of a gastropod hemocyanin offers a possible mechanism for cooperativity. Biochemistry, 42(21): 6341–6346. Doi: 10.1021/bi020672x |
| Pho L N, Leachman S A, 2010. Genetics of pigmentation and melanoma predisposition. Giornale italiano di Dermatologia e Venereologia, 145(1): 37–45. |
| Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O, Kinch L, Sheffler W, Kim B H, Das R, Grishin N V, Baker D, 2009. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins, 77(S9): 89–99. Doi: 10.1002/prot.v77.9s |
| Roy A, Kucukural A, Zhang Y, 2010. I-TASSER:a unified platform for automated protein structure and function prediction. Nature Protocols, 5(4): 725–738. Doi: 10.1038/nprot.2010.5 |
| Samata T, Hayashi N, Kono M, Hasegawa K, Horita C, Akera S, 1999. A new matrix protein family related to the nacreous layer formation of Pinctada fucata. FEBS Letters, 462(1-2): 225–229. Doi: 10.1016/S0014-5793(99)01387-3 |
| Sendovski M, Kanteev M, Ben-Yosef V S, Adir N, Fishman A, 2011. First structures of an active bacterial tyrosinase reveal copper plasticity. Journal of Molecular Biology, 405(1): 227–237. Doi: 10.1016/j.jmb.2010.10.048 |
| Shen X, Belcher A M, Hansma P K, Stucky G D, Morse D E, 1997. Molecular cloning and characterization of lustrin A, a matrix protein from shell and pearl nacre of Haliotis rufescens. J Biol Chem., 272(51): 32472–32481. Doi: 10.1074/jbc.272.51.32472 |
| Shuster Ben-Yosef V, Sendovski M, Fishman A, 2010. Directed evolution of tyrosinase for enhanced monophenolase/diphenolase activity ratio. Enzyme and Microbial Technology, 47(7): 372–376. Doi: 10.1016/j.enzmictec.2010.08.008 |
| Slominski A, Tobin D J, Shibahara S, Wortsman J, 2004. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiological Reviews, 84(4): 1155–1228. Doi: 10.1152/physrev.00044.2003 |
| Smith B L, Schäffer T E, Viani M, Thompson J B, Frederick N A, Kindt J, Belcher A, Stucky G D, Morse D E, Hansma P K, 1999. Molecular mechanistic origin of the toughness of natural adhesives, fibres and composites. Nature, 399(6738): 761–763. Doi: 10.1038/21607 |
| Söding J, 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics, 21(7): 951–960. Doi: 10.1093/bioinformatics/bti125 |
| Solomon E I, Heppner D E, Johnston E M, Ginsbach J W, Cirera J, Qayyum M, Kieber-Emmons M T, Kjaergaard C H, Hadt R G, Tian L, 2014. Copper active sites in biology. Chemical Reviews, 114(7): 3659–3853. Doi: 10.1021/cr400327t |
| Suhre M H, Gertz M, Steegborn C, Scheibel T, 2014. Structural and functional features of a collagen-binding matrix protein from the mussel byssus. Nature Communications, 5(5): 3392–3392. |
| Takgi R, Miyashita T, 2013. A cDNA cloning of a novel alphaclass tyrosinase of Pinctada fucata:its expression analysis and characterization of the expressed protein. Enzyme Research, 2014(2): 780549–780549. |
| Tamura K, Stecher G, Peterson D, Filipski A, Kumar S, 2013. MEGA6:molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30(12): 2725–2729. Doi: 10.1093/molbev/mst197 |
| True J R, 2003. Insect melanism:the molecules matter. Trends in Ecology & Evolution, 18(12): 640–647. |
| Yan F, Luo S J, Jiao Y, Deng Y W, Du X D, Huang R L, Wang Q H, Chen W Y, 2014. Molecular characterization of the BMP7 gene and its potential role in shell formation in Pinctada martensii. International Journal of Molecular Sciences, 15(11): 21215–21228. Doi: 10.3390/ijms151121215 |
| Yano M, Nagai K, Morimoto K, Miyamoto H, 2006. Shematrin:a family of glycine-rich structural proteins in the shell of the pearl oyster Pinctada fucata. Comparative Biochemistry and Physiology Part B:Biochemistry and Molecular Biology, 144(2): 254–262. Doi: 10.1016/j.cbpb.2006.03.004 |
| Yu J, Wei W, Danner E, Ashley R K, Israelachvili J N, Waite J H, 2011. Mussel protein adhesion depends on interprotein thiol-mediated redox modulation. Nature Chemical Biology, 7(9): 588–590. Doi: 10.1038/nchembio.630 |
| Zhang C, Xie L P, Huang J, Chen L, Zhang R Q, 2006. A novel putative tyrosinase involved in periostracum formation from the pearl oyster (Pinctada fucata). Biochemical and Biophysical Research Communications, 342(2): 632–639. Doi: 10.1016/j.bbrc.2006.01.182 |
| Zhang G F, Fang X D, Guo X M, et al, 2012. The oyster genome reveals stress adaptation and complexity of shell formation. Nature, 490(7418): 49–54. Doi: 10.1038/nature11413 |
| Zhao X X, Wang Q H, Jiao Y, Huang R L, Deng Y W, Wang H, Du X D, 2012. Identification of genes potentially related to biomineralization and immunity by transcriptome analysis of Pearl Sac in Pearl Oyster Pinctada martensii. Marine Biotechnology, 14(6): 730–739. Doi: 10.1007/s10126-012-9438-3 |
| Zhou Z, Ni D J, Wang M Q, Wang L L, Shi X W, Yue F, Liu R, Song L S, 2012. The phenoloxidase activity and antibacterial function of a tyrosinase from scallop Chlamys farreri. Fish & Shellfish Immunology, 33(2): 375–381. |