 2020, Vol. 38
2020, Vol. 38Institute of Oceanology, Chinese Academy of Sciences
Article Information
- XUE Dongxiu, YANG Qiaoli, ZONG Shaobing, GAO Tianxiang, LIU Jinxian
- Genetic variation within and among range-wide populations of three ecotypes of the Japanese grenadier anchovy Coilia nasus with implications to its conservation and management
- Journal of Oceanology and Limnology, 38(3): 851-861
- http://dx.doi.org/10.1007/s00343-019-9091-z
Article History
- Received Apr. 9, 2019
- accepted in principle Jun. 17, 2019
- accepted for publication Aug. 15, 2020




2 Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China;
3 Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China;
4 University of Chinese Academy of Sciences, Beijing 100049, China;
5 School of Fisheries, Zhejiang Ocean University, Zhoushan 316022, China
Conservation of life history variations is an important consideration for many species with trade-offs in migratory characteristics (Nichols et al., 2016). The Japanese grenadier anchovy, Coilia nasus, is widely distributed along the coasts of China, the western coasts of the Korean Peninsula, the Ariake Sea of Japan, and the Changjiang (Yangtze) River and other major rivers in China (Whitehead et al., 1988). Based on the previous morphological and ecological studies of this species, three ecotypes of grenadier anchovy were identified: anadromous, landlocked, and freshwater resident populations (Cheng, 2011). Anadromous grenadier anchovy spends adult life in the ocean and breeds in freshwater, whereas landlocked and freshwater resident grenadier anchovy inhabit only freshwater (Yuan et al., 1980; Cheng, 2011). In the past, the freshwater resident grenadier anchovy used to be named as Coilia brachygnathus, which resides Changjiang River and its adjoining lakes. The jaw length of the freshwater-resident ecotype (shortjaw tapertail anchovy) is shorter than those of the anadromous and landlocked ecotype. The landlocked grenadier anchovy used to be named as Coilia nasus taihuensis, which inhabits the lakes isolated to the Changjiang River and Huaihe River (such as Taihu Lake, Chaohu Lake, and Hongze Lake). The landlocked grenadier anchovy is a typical example of a human-induced alteration of the life history of the anadromous fishes (Yang et al., 2006). The analysis based on the microchemistry patterns of element strontium and calcium in otoliths also supported that grenadier anchovy individuals in Taihu Lake are landlocked (Yang et al., 2006). There seems to be no evidence of large-scale geological activity that would have blocked spawning migrations or isolated inland lakes (e.g. Taihu Lake, Chaohu Lake, and Hongze Lake) (Xue et al. 2019). However, hydraulic facilities are a ubiquitous feature of Taihu Lake, Chaohu Lake, and Hongze Lake undoubtedly disrupted the migrations of anadromous fishes, including grenadier anchovies, which migrate up the lakes outlet upon which some hydraulic facilities (e.g. Chaohu Sluice, Yuxi Sluice, Taipu Sluice, Sanhe Sluice, and Erhe Sluice) were constructed since 1950s. As a lucrative commercial fishery, natural anadromous populations have been declining dramatically in China due to over-exploitation and deterioration of the coastal environment and spawning grounds (Zhang et al., 2005). When population sizes reduced to a level, inbreeding and loss of genetic diversity occur and may result in extinction of local populations (Reed et al., 2003). To protect this species, national aquatic germplasm resources conservation area of China of grenadier anchovy have been built in Changjiang River.
Genetic diversity and population genetic structure are fundamental to the understanding of the evolutionary and ecological process that influence biodiversity and provide explicit framework for conservation of species (Frankham et al., 2009). Fisheries management can benefit from considering population genetic diversity and differentiation as well as how biological characteristics and environmental factors influence population connectivity. Most of the previous studies on C. nasus were mainly focused on the genetic relationships among the three ecotypes of C. nasus based on mitochondrial DNA markers, and only a small number of populations were analyzed (Tang et al., 2007; Cheng et al., 2008; Liu et al., 2012, 2014; Yang et al., 2017). Also, previous findings on the genetic relationships among different ecotypes were somewhat conflicted (Yang, 2012; Liu et al., 2014; Yang et al., 2017). Yet, little is known about a current genetic background of C. nasus across its distribution range, which may become an impediment to the ongoing fisheries management and conservation of C. nasus.
In the present study, two anonymous fragments flanking C. nasus (AGAT)N microsatellite loci were tested for utility as 'putatively neutral' markers for inference of genetic variation within and among 17 populations of C. nasus from China and one population from the Ariake Sea in Japan. The aim of this study was to estimate the levels of genetic diversity within each population and to infer population structure among the 18 populations. The results would provide useful information for proper scientific and judicious management actions to ensure sustainability of C. nasus populations.
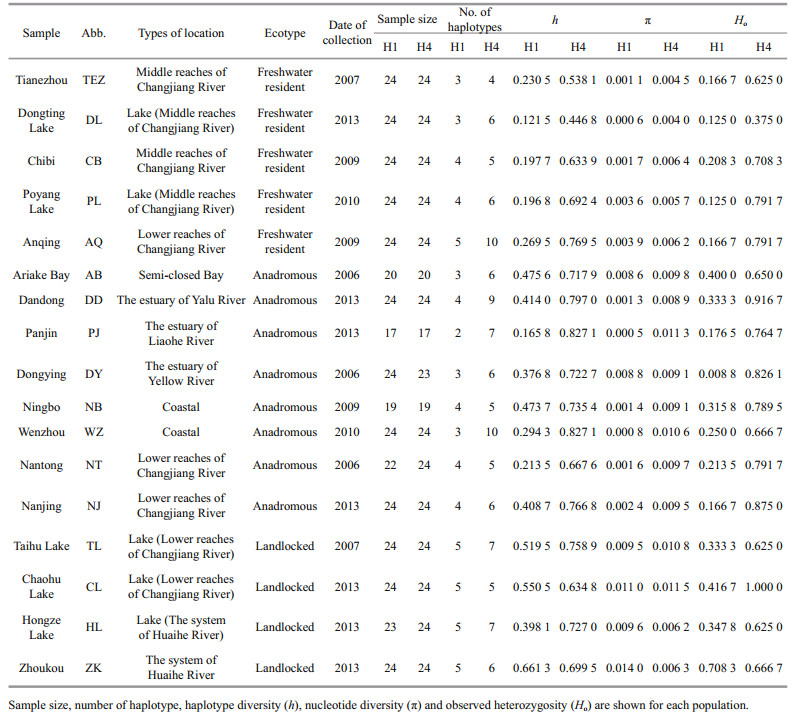
2 MATERIAL AND METHOD 2.1 Sample collection and DNA extractionCoilia nasus specimens were collected from 18 sites in China and Japan (17 to 24 individuals per site) during 2006–2013 (Table 1). Genomic DNA was extracted from muscle tissue by proteinase K digestion followed by a standard phenol-chloroform method.
PCR primers were designed to amplify 361 bp and 264 bp anonymous fragments flanking C. nasus (AGAT) N microsatellite loci (Locus H1 and Locus H4, CnasH1F: 5′-GTC CGT AGT AAC CGA AAT TGT G, CnasH1R: 5′-TCC CTC GAC TGT CAT ATC ATG T; CnasH4F: 5′-CCA GTC ATG AGT ATG ACC TC, CnasH4R: 5′-CCT CTT TGT ATT ATT GGG AA). The nuclear sequences were amplified for 17–24 individuals per site (Table 1). PCRs were performed in 25 μL volumes followed by Wilson and Veraguth (2010). PCR amplification included 5 min initial denaturation at 94℃, and 35 cycles of 94℃ (40 s), 56℃ (H1) or 53℃ (H4) (50 s) and 72℃ (1 min), and a final extension at 72℃ for 10 min. PCR products were sequenced on an ABI 3730XL automated sequencer with both forward and reverse primers (Life Technologies Biotechnology Co., China).
Allelic sequences were inferred using a two-step procedure (Wilson and Veraguth, 2010). The first step involved the statistical inference of allelic phase using the default settings of PHASE V2.1 (http://stephenslab.uchicago.edu/software.html#phase). This method provides accurate measures in statistically estimated alleles that are well-represented in a diploid data set, but may be uncertain about reconstructing allelic phase for unique genotypes (Harrigan et al., 2008; Wilson and Veraguth, 2010). Individuals for which allelic phase was uncertainty were reamplified and cloned into TA cloning vectors (TaKaRa) following the manufacturer's protocol. Three clones were picked randomly and sequenced for each individual, allowing the direct determination of allelic identify.
2.3 Statistical analysisObserved heterozygosity of H1 and H4 alleles was calculated using Arlequin v3.5 (Excoffier and Lischer, 2010). Molecular diversity indices such as haplotype diversity (h), nucleotide diversity (π), number of polymorphic sites and haplotypes were obtained using the program Arlequin v3.5 (Excoffier and Lischer, 2010).
Phylogenetic relationships among haplotypes were constructed using MrBayes 3.1.2 (Ronquist and Huelsenbeck, 2003). A MrBayes setting for the best fit model (HKY+I+G) was selected by the hierarchical likelihood ratio tests with MrModeltest 2.3 (Nylander, 2004) in conjunction with PAUP 4.0b10 (Swofford, 2002). Markov chains were run for 107 generations (the average SD of split frequencies < 0.01) with four chains starting from a random tree. Sampling frequency was set at 100 generations and the first 7 500 samples (25%) were excluded as burn-in (Shikano et al., 2010). Genealogical relationships among haplotypes for both the H1 and H4 gene were further assessed using a minimum spanning tree constructed by Arlequin V3.5 (Excoffier and Lischer, 2010).
Population structure was evaluated with AMOVA analyses that incorporate haplotype frequency and sequence divergence among haplotypes (Excoffier et al., 1992). We conducted AMOVA analyses with two groups: the freshwater resident group, anadromous and landlocked group. The significance of covariance component associated with the different possible levels of genetic structure was tested using 10 000 permutations. In addition, the fixation index ΦST was used to compare the genetic divergence values between populations, and statistical significance was evaluated by 10 000 permutations for each pairwise comparison (Excoffier et al., 1992). All the population divergence calculations were performed in Arlequin V3.5 (Excoffier and Lischer, 2010).
3 RESULT 3.1 Genetic diversitySequence comparison for the 361 bp of the H1 nuclear fragment revealed 19 distinct haplotypes defined by 27 polymorphic sites. A single-base deletion in Locus H1 was common in freshwater resident individuals, but rare in the rest individuals. For H1 fragment, the number of haplotypes ranged from 2 (Panjin) to 5 (Anqing, Taihu Lake, Chaohu Lake, Hongze Lake, and Zhoukou), haplotype diversity (h) ranged from 0.121 5 (Dongting Lake) to 0.661 3 (Zhoukou), nucleotide diversity (π) ranged from 0.000 6 (Dongting Lake) to 0.014 0 (Zhoukou), and the observed heterozygosity (Ho) ranged from 0.125 0 (Dongting Lake and Poyang Lake) to 0.708 3 (Zhoukou) (Table 1). For the 264 bp of the H4 nuclear fragment, 19 distinct haplotypes were defined by 15 polymorphic sites. The number of haplotypes ranged from 4 (Tianezhou) to 10 (Anqing and Wenzhou), the value of h ranged from 0.446 8 (Dongting Lake) to 0.827 1 (Panjin and Wenzhou), the value of π varied from 0.004 0 (Dongting Lake) to 0.011 5 (Chaohu Lake), and Ho ranged from 0.375 0 (Dongting Lake) to 1.000 0 (Chaohu Lake) for H4 fragment (Table 1).
3.2 Population structureFor the H1 fragment, pairwise FST values ranged from -0.021 3 between Ariake Bay and Dongying to 0.980 9 between Dongting Lake and Panjin (Table 2). All of the five freshwater resident populations were significantly different from the rest 13 populations after a Benjamini-Yekutieli correction (P < 0.008 9). However, genetic differences among the five freshwater resident populations were non-significant (P > 0.008 9). The genetic divergences were significant between Ariake Bay in Japan and five anadromous populations in China (Dandong, Panjin, Wenzhou, Nantong and Nanjing). The four landlocked populations (Taihu Lake, Chaohu Lake, Hongze Lake and Zhoukou) were significant significantly different from six anadromous populations except for Dongying population. Dongying population was significantly different from five anadromous populations (Dandong, Panjin, Wenzhou, Nantong, and Nanjing) and one landlocked population (Zhoukou).

|
For the H4 fragment, pairwise FST values ranged from -0.020 3 between Panjin and Wenzhou to 0.543 5 between Chibi and Ningbo (Table 2). All of the five freshwater resident populations were significantly different from the rest 13 populations after a Benjamini– Yekutieli correction (P < 0.008 9) except for Poyang Lake & Hongze Lake, Anqing & Hongze Lake, and Anqing & Zhoukou. The two landlocked populations (Hongze Lake and Anqing) of Huaihe River were significantly divergent with the eight anadromous populations and the other two landlocked populations (Taihu Lake and Chaohu Lake) (P < 0.008 9).
Using the haplotypes of H1 fragment, two distinct lineages (labelled A and B) were revealed by Bayesian inference of phylogeny under the HKY+I model (Fig. 1). The lineage A dominated the five freshwater resident populations. This result was further supported by the minimum spanning tree for H1 fragment (Fig. 2). The minimum spanning tree for H1 fragment indicated that the two lineages were separated from each other by seven mutational steps (Fig. 2). There were strong geographical differences in haplotype frequencies of H1 fragment. Three haplotypes (HAP1, HAP4 and HAP5) were common in the five freshwater resident populations. However, the HAP1 was also found in four landlocked populations (Hongze Lake, Zhoukou, Chaohu Lake, and Taihu Lake) and two anadromous populations (Dongying and Ariake Bay).

|
| Fig.1 Bayesian tree for H1 haplotypes, as constructed under the HKY+I substitution model with MRBAYES Numbers above branches show posterior probabilities of nodes. |

|
| Fig.2 Minimum spanning tree showing genetic relationship among H1 haplotypes |
Additionally, three common haplotypes (HAP2, HAP3, and HAP10) were observed in the rest 13 populations from Huaihe River, Lower Changjiang River and coastal populations. HAP2 was also existed at low frequency in two freshwater resident populations (Poyang Lake and Anqing) (Fig. 3).

|
| Fig.3 Pie charts show the frequencies of H1 haplotypes among populations of C. nasus |
For the H4 fragment, the phylogenetic tree was obtained by Bayesian analysis under the F81+I model (Fig. 4). The minimum spanning tree for this fragment was defined by five common haplotypes (HAP1, 2, 4, 6, and 7, Fig. 5), one of which (HAP7) was mainly restricted to the five freshwater resident populations, and one landlocked population from Chaohu Lake. HAP6 was found at high frequency in the seven anadromous populations in China and two landlocked population (Taihu Lake and Chaohu Lake) from the Lower Changjiang River (Fig. 6). HAP2 was found at high frequency in one freshwater resident population (Anqing), seven anadromous populations in China and three landlocked populations (Taihu Lake, and Hongze Lake, and Zhoukou) (Fig. 6).

|
| Fig.4 Bayesian tree for H4 haplotypes, as constructed under the F81+I substitution model with MRBAYES Numbers above branches show posterior probabilities of nodes. |

|
| Fig.5 Minimum spanning tree showing genetic relationship among H4 haplotypes |

|
| Fig.6 Pie charts show the frequencies of H4 haplotypes among populations of C. nanus |
Based on the results of pairwise FST values, Bayesian trees and minimum spanning trees, AMOVA analyses were conducted with two groups: freshwater resident group, and (anadromous & landlocked) group. For the H1 fragment, the AMOVA analysis revealed that most of the genetic variation was distributed between two groups (77.43%, P=0.00). Smaller but significant amount of variances (P=0.00) were also found within populations (19.77%) and among populations within groups (2.79%). For the H4 fragment, AMOVA analysis revealed that 72.98% of the genetic diversity was found within populations (P=0.00). Smaller but significant amount of variances (P=0.00) were also found between two groups (21.03%) and among populations within groups (5.99%).
4 DISCUSSIONThe level of genetic diversity of C. nasus was characterized by moderate haplotype diversity (10 of 18 populations has low values based on H1 fragment) and low nucleotide diversity. In addition, the relative low haplotype diversity, nucleotide diversity, and observed heterozygosity based on both H1 and H4 fragments were found in the five freshwater resident populations from Middle Changjiang River. These results were consistent with the conclusions of a previous survey of microsatellite variation in marine, freshwater and anadromous fishes reported by DeWoody and Avise (2000). Anadromous fish populations tend to display higher levels of genetic diversity than do freshwater fishes, which probably are attributable in part to differences in evolutionarily effective population sizes and chances for admixture of typifying species inhabiting these realms (DeWoody and Avise, 2000).
The results of AMOVA, pairwise FST values, the Bayesian phylogenetic tree and haplotypes networks revealed high levels of genetic divergence between freshwater resident group and (anadromous & landlocked) group. Additionally, strong allele frequency changes of two nuclear fragments and high pairwise FST values indicated limited genetic exchange between freshwater resident populations and anadromous middle and lower reaches of Changjiang River populations in the absence of contemporary dispersal barriers. Similar genetic divergences between populations in the middle and lower reaches of the Changjiang River have also been reported in some other aquatic organisms (Lu et al., 1997; Feng et al., 2008; Zhao et al., 2008). These concordant patterns reveal that historical geographical factors greatly influenced the evolutionary genetic structure of aquatic organisms in the middle and lower reaches of the Changjiang River (Liu et al., 2007). During the last Pleistocene glaciation, the water level of the Changjiang River dropped more than 20 m with the fall of sea levels (Yang, 1986). Some geological researches have proved that the Changjiang River was an inland river and did not flow into East China Sea during last glacial maximum for the impacts of arid climate, reduced rainfall and desertification (Xiao et al., 2003). Thus, some populations of C. nasus were probably isolated in main deep-water channel of the Changjiang River in Pleistocene glaciations and became freshwater resident populations eventually (Xue et al., 2019; Yang, 2012).
Besides historical geographical factors, contemporary anthropogenic factors also play an important role in shaping the present-day genetic structure. The landlocked ecotype of C. nasus was formally described in 1976 (Yuan et al., 1976, Yang et al., 2006). In recent years, human activities have made a drastic impact on the Changjiang River and Huaihe River, and many hydraulic facilities have been constructed in Chaohu Lake, Taihu Lake, and Huaihe River basin, including sluices and dams (Yang et al., 2006; Hu et al., 2008). Anadromous C. nasus have become the landlocked individuals when their migration waterway was blocked. Such rapid phenotypic evolution was common in fishes, such as in guppies, rainbow trout and stickleback (Messer et al., 2016). One prominent example is the life history traits change of marine sticklebacks to landlocked ecotype, which has occurred within the last 50 years after an earthquake created multiple freshwater ponds (Lescak et al., 2015). In the present study, pairwise FST values showed significant genetic differentiation (pairwise FST values) observed between the four landlocked populations and the six Chinese anadromous populations except Dongying population based on the H1 fragment. For the H4 fragment, the two landlocked populations (Hongze Lake and Anqing) of Huaihe River were significantly divergent with the eight anadromous populations and the other two landlocked populations (Taihu Lake and Chaohu Lake). However, the haplotypes frequencies of nuclear H1 fragment showed HAP2 dominated the four landlocked populations and the nine anadromous populations. For the H4 fragment, HAP2 and HAP6 dominated the four landlocked populations and the nine anadromous populations. In this case, the genetic differentiation between landlocked and anadromous populations could reflect the recency of the formation of all the landlocked populations and genetic bottleneck.
5 CONCLUSIONIn summary, our range-wide examination of genetic variation among populations of C. nasus suggested that the relative influence of geographic isolation, anthropogenic factors, biological characteristics, and historical events on contemporary patterns vary spatially. The results of our study detected significant genetic structure of C.nasus, which might arise from current geographic segregation, different life-history strategy, and historical geographical factors. C. nasus freshwater resident populations reflected a unique genetic cluster and relative lower genetic diversity. Patterns of genetic differentiation observed on the landlocked populations (Chaohu Lake, Taihu Lake, Hongze Lake, and Zhoukou) were probably the genetic effects of hydraulic facilities in lower Changjiang River and Huaihe River. Populations with demographic independence should be monitored and managed separately as management units (MUs). MUs are the basis for the short-term management of populations and are crucial for monitoring the effects of human activity on species diversity (Palsbøll et al., 2007). The most obvious conservation finding from our results is the identification of at least three MUs: the freshwater resident populations, the landlocked populations, and the anadromous populations. The results in the present study provide complementary information for conducting genetic stock identification and setting stock specific management plans for C.nasus in China. Our study demonstrated the utility of putatively neutral nuclear data to inform conservation in highly exploited species and improve stock-specific management in China.
6 DATA AVAILABILITY STATEMENTSequence data that support the findings of this study are available from GenBank under the accessions Nos. MK777930–MK777967.
Cheng Q Q, Cheng H P, Wang L, Zhong Y, Lu D R. 2008. A preliminary genetic distinctness of four Coilia fishes(Clupeiformes:Engraulidae) inferred from mitochondrial DNA sequences. Russian Journal of Genetics, 44(3): 339-343.
DOI:10.1134/S1022795408030150 |
Cheng W X. 2011. The Research of Some Phenotypic Differences from Different Ecotypes of Coilia nasus from Yangtze River. Shanghai Ocean University, Shanghai.
(in Chinese with English abstract)
|
DeWoody J A, Avise J C. 2000. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. Journal of Fish Biology, 56(3): 461-473.
DOI:10.1111/j.1095-8649.2000.tb00748.x |
Excoffier L, Lischer H E L. 2010. Arlequin suite ver 3.5:a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3): 564-567.
DOI:10.1111/j.1755-0998.2010.02847.x |
Excoffier L, Smouse P E, Quattro J M. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes:application to human mitochondrial DNA restriction data. Genetics, 131(2): 479-491.
|
Feng J B, Sun Y N, Cheng X, Li J L. 2008. Sequence analysis of mitochondrial COI gene of Macrobrachium nipponense from the five largest freshwater lakes in China. Journal of Fisheries of China, 32(4): 517-525.
|
Frankham R, Ballou J D, Briscoe D A. 2009. Introduction to Conservation Genetics. Cambridge University Press, Cambridge, UK. 704p.
|
Harrigan R J, Mazza M E, Sorenson M D. 2008. Computation vs. cloning:evaluation of two methods for haplotype determination. Molecular Ecology Resources, 8(6): 1 239-1 248.
|
Hu W W, Wang G X, Deng W, Li S N. 2008. The influence of dams on ecohydrological conditions in the Huaihe River basin, China. Ecological Engineering, 33(3-4): 233-241.
DOI:10.1016/j.ecoleng.2008.04.003 |
Lescak E A, Bassham S L, Catchen J, Gelmond O, Sherbick M L, von Hippel F A, Cresko W A. 2015. Evolution of stickleback in 50 years on earthquake-uplifted islands. Proceedings of the National Academy of Sciences of the United States of America, 112(52): E7 204-E7 212.
DOI:10.1073/pnas.1512020112 |
Liu D, Guo H Y, Tang W Q, Yang J Q. 2012. Comparative evolution of S7 intron 1 and ribosomal internal transcribed spacer in Coilia nasus (Clupeiformes:Engraulidae). International Journal of Molecular Sciences, 13(3): 3 085-3 100.
DOI:10.3390/ijms13033085 |
Liu D, Li Y Y, Tang W Q, Yang J Q, Guo H Y, Zhu G L, Li H H. 2014. Population structure of Coilia nasus in the Yangtze River revealed by insertion of short interspersed elements. Biochemical Systematics and Ecology, 54: 103-112.
DOI:10.1016/j.bse.2013.12.022 |
Liu J X, Gao T X, Wu S F, Zhang Y P. 2007. Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus(Temminck & Schlegel, 1845). Molecular Ecology, 16(2): 275-288.
DOI:10.1111/j.1365-294X.2006.03140.x |
Lu G Q, Li S F, Bernatchez L. 1997. Mitochondrial DNA diversity, population structure, and conservation genetics of four native carps within the Yangtze River, China. Canadian Journal of Fisheries and Aquatic Sciences, 54(1): 47-58.
DOI:10.1139/f96-266 |
Messer P W, Ellner S P, Hairston N G. 2016. Can population genetics adapt to rapid evolution?. Trends in Genetics, 32(7): 408-418.
DOI:10.1016/j.tig.2016.04.005 |
Nichols K M, Kozfkay C C, Narum S R. 2016. Genomic signatures among Oncorhynchus nerka ecotypes to inform conservation and management of endangered Sockeye Salmon. Evolutionary Applications, 9(10): 1 285-1 300.
DOI:10.1111/eva.12412 |
Nylander J A A. 2004. MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University.
|
Palsbøll J P, Bérubé M, Allendorf F W. 2007. Identification of management units using population genetic data. Trends in Ecology & Evolution, 22(1): 11-16.
|
Reed D H, Lowe E H, Briscoe D A, Frankham R. 2003. Inbreeding and extinction:Effects of rate of inbreeding. Conservation Genetics, 4(3): 405-410.
DOI:10.1023/A:1024081416729 |
Ronquist F, Huelsenbeck J P. 2003. MRBAYES 3:Bayesian phylogenetic inference under mixed models. Bioinformatics, 19(12): 1 572-1 574.
DOI:10.1093/bioinformatics/btg180 |
Shikano T, Shimada Y, Herczeg G, Merilä J. 2010. History vs. habitat type:explaining the genetic structure of European nine-spined stickleback (Pungitius pungitius) populations.Molecular Ecology, 19(6): 1 147-1 161.
|
Swofford D L. 2002. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), version 4.0b10. Sinauer Associates, Sunderland, MA.
|
Tang W Q, Hu X L, Yang J Q. 2007. Species validities of Coilia brachygnathus and C. nasus taihuensis based on sequence variations of complete mtDNA control region.Biodiversity Science, 15(3): 224-231.
|
Whitehead P J P, Nelson G J, Wongratana T. 1988. FAO species catalogue. Clupeoid fishes of the world (Suborder Clupeoidei). An annotated and illustrated catalogue of the herrings, sardines, pilchards, sprats, shads, anchovies and wolf-herrings. Part 2:Engraulididae. FAO Fisheries Synopsis, 125(7): 305-579.
|
Wilson A B, Veraguth I E. 2010. The impact of Pleistocene glaciation across the range of a widespread European coastal species. Molecular Ecology, 19(20): 4 535-4 553.
DOI:10.1111/j.1365-294X.2010.04811.x |
Xiao S B, Huang P, Wan S M, Li A C. 2003. Entering sea history of the Yangtze River during the last pleniglacial stage. Journal of the University of Petroleum, China, 27(6): 125-130.
(in Chinese with English abstract) |
Xue D X, Yang Q L, Li Y L, Zong S B, Gao T X, Liu J X. 2019. Comprehensive assessment of population genetic structure of the overexploited Japanese grenadier anchovy(Coilia nasus):implications for fisheries management and conservation. Fisheries Research, 213: 113-120.
DOI:10.1016/j.fishres.2019.01.012 |
Yang D Y. 1986. The paleoenvironment of the mid-lower regions of Changjiang in the full-glacial period of late Pleistocene. Acta Geographica Sinica, 53(4): 302-310.
(in Chinese with English abstract) |
Yang J Q, Hsu K C, Zhou X D, Kuo P H, Lin H D, Liu D, Bao B L, Tang W Q. 2017. New insights on geographical/ecological populations within Coilia nasus (Clupeiformes:Engraulidae) based on mitochondrial DNA and microsatellites. Mitochondrial DNA Part A:DNA Mapping, Sequencing, and Analysis, 29(1): 158-164.
|
Yang J, Arai T, Liu H, Miyazaki N, Tsukamoto K. 2006. Reconstructing habitat use of Coilia mystus and Coilia ectenes of the Yangtze River estuary, and of Coilia ectenes of Taihu Lake, based on otolith strontium and calcium. Journal of Fish Biology, 69(4): 1 120-1 135.
DOI:10.1111/j.1095-8649.2006.01186.x |
Yang Q L. 2012. Phylogenetic Analysis of Genus Coilia in China and Molecular Phylogeography of C. nasus and C.mystus. Ocean University of China, Qingdao.
(in Chinese with English abstract)
|
Yuan C M, Lin J B, Qin A L, Liu R H. 1976. Historical and present taxonomic status about the genus Coilia in China. Journal of Nanjing University, (2): 1-12.
(in Chinese with English abstract) |
Yuan C M, Qin A L, Liu R H, Lin J B. 1980. On the classification of the anchovies, Coilia, from the lower Yangtze River and the southeast coast of China. Journal of Nanjing University, (3): 67-77.
(in Chinese with English abstract) |
Zhang M Y, Xu D P, Liu K, Shi W G. 2005. Studies on biological characteristics and change of resource of Coilia nasus Schlegel in the lower reaches of the Yangtze River. Resources and Environment in the Yangtze Basin, 14(6): 694-698.
(in Chinese with English abstract) |
Zhao L, Zhang J, Liu Z J, Funk S M, Wei F W, Xu M Q, Li M. 2008. Complex population genetic and demographic history of the Salangid, Neosalanx taihuensis, based on cytochrome b sequences. BMC Evolutionary Biology, 8(1): 201.
DOI:10.1186/1471-2148-8-201 |