 2017, Vol. 35
2017, Vol. 35Institute of Oceanology, Chinese Academy of Sciences
Article Information
- ZHANG Hui(张辉), ZHAI Yuxiu(翟毓秀), YAO Lin(姚琳), JIANG Yanhua(江艳华), LI Fengling(李风铃)
- Comparative transcriptomics reveals genes involved in metabolic and immune pathways in the digestive gland of scallop Chlamys farreri following cadmium exposure
- Chinese Journal of Oceanology and Limnology, 35(3): 603-612
- http://dx.doi.org/10.1007/s00343-017-5305-4
Article History
- Received Dec. 4, 2015
- accepted in principle Mar. 4, 2016




2 Key Laboratory of Testing and Evaluation for Aquatic Product Safety and Quality, Ministry of Agriculture, China;
3 Laboratory of Quality and Safety Risk Assessment for Aquatic Products (Qingdao), Ministry of Agriculture, China
The scallop Chlamys farreri is an important commercial species that is cultivated widely by the rope-caging method in the northern coastal provinces of China. In recent years, anthropogenic activities, such as high summer temperatures and environmental pollution, have damaged scallop aquaculture (Nagashima et al., 2005). Many chemical contaminants and heavy metals are now considered to be harmful to aquaculture environments (Haynes and Johnson, 2000; Torres et al., 2008). Chlamys farreri are filter feeders that have been proved to be effective concentrators of heavy metals. Previous related studies on mollusk have focused mainly on the effects on antioxidant enzyme activity (Shan et al., 2011), lipid peroxidation (Lukyanova and Khotimchenko, 1995), testing bioaccumulation in tissues (Chelomin et al., 1995), DNA strand breakage (Slobodskova et al., 2012), and the discovery of genetic markers (Sato et al., 2005; Wang et al., 2012). However, these studies were limited due to the lack of genomic resources such as genome and transcriptomic sequences. Understanding the mechanism of detoxification in C. farreri is a high priority for the aquaculture industry. In order to achieve this goal, it is necessary to obtain a global understanding of the transcriptomic profile of C. farreri.
Cadmium (Cd) is a heavy metal that can be accumulated easily and it is one of the top 17 types of poison in contaminated food identified by the World Health Organization. Cd pollution can be accumulated through the food chain, and be enriched and amplified. According to an ecological assessment conducted from 2001 to 2005, Cd pollution is a serious problem in Bohai Bay, northeast China (Peng et al., 2009). Cd pollution has become a major problem that affects the quality and safety of aquatic products, especially scallops. Therefore, it is necessary to understand the toxicological mechanism in C. farreri.
In recent years, next-generation sequencing technologies have been used successfully for gene expression analysis, identification of differentially expressed genes, and discovery of novel transcripts (Garber et al., 2011) of genome reference-free species and non-model organisms. For example, RNA-seq has been applied in various invertebrates, such as Eriocheir sinensis (He et al., 2012; Ou et al., 2012), Bactrocera dorsalis (Hsu et al., 2012), and Pinctada martensii (Zhao et al., 2012) and other bivalves (Gavery and Roberts, 2012; Meng et al., 2014).
In the present study, we analyzed the digestive gland of C. farreri exposed to Cd based on Illumina sequencing and bioinformatics methods. The main objective of this study was to annotate functional genes related to Cd accumulation and identify potential molecular mechanisms involved in metabolic and immune pathways in C. farreri.
2 MATERIAL AND METHOD 2.1 Experimental scallopAdult C. farreri scallops were collected from a commercial farm in Qingdao, China. Housing and care of the scallops and the collection of tissue samples were conducted in accordance with the International Guiding Principles for Biomedical Research Involving Animals. Only healthy scallops of a homogeneous size were used. The scallops were kept in running aerated sea water (salinity 30) at 20±1℃ that was pumped from Aoshanwei seaside of Qingdao and filtered prior to use. All of the seawater was changed daily and the scallops were fed with dried powder of Spirulina platensis (3 g/cm3 water per day).
2.2 Cadmium exposure and RNA preparationThe scallops were divided into two groups: one group was the control, and the other group was exposed to CdCl2.2.5 H2O. The final Cd2+ concentration reached 0.2 mg/L, which is 40 times of the standard formulated by 'Water Quality Standard for Fisheries of China' (Cd2+≤0.005 mg/L). No mortality was observed during the experimental period. After 15 days exposure, tissues of the digestive glands from five scallops in the control and five scallops in the Cd-treated groups were collected for RNA-seq. Two independent biological replicates (Cd-treated groups: XC1 and XC2; control groups: XD1 and XD2) were prepared for each group. Total RNA was isolated from tissues using TRIzol (TaKaRa, Dalian, China) according to the manufacturer's protocol. The quality and quantity of RNA was tested on an Agilent 2100 Bioanalyzer (Agilent Technologies) and a NanoDrop spectrophotometer (Thermo Scientific, USA), respectively. The total RNA was then treated with DNase Ⅰ (Ambion, USA) for 1 h at 37℃ to remove residual DNA from the total RNA.
2.3 cDNA library construction and RNA-seqOne paired-end (PE) cDNA library was generated from the total RNA of the digestive glands. Briefly, oligo (dT) beads were used to isolate poly (A) mRNA (mixture). The first-strand cDNA was synthesized using a random hexamer primer and reverse transcriptase, and the second-strand cDNA using DNA polymerase Ⅰ (TaKaRa) and RNase H (TaKaRa). The gel-purified cDNA product was modified using a TruSeqTM DNA Sample Prep Kit-Set A (Illumina) for sequencing, and then PCR amplified using a TruSeq Paired-End Cluster Kit (Illumina).
2.4 Analysis of Illumina sequencing resultsThe cDNA library was sequenced on the Illumina platform (GA Ⅱ). The raw data generated were assembled into non-redundant consensus unigenes sequences using two assemblers for very short reads: Velvet (version 0.7.62; http://www.ebi.ac.uk/zerbino/ velvet/) (Zerbino and Birney, 2008) and Oases (version 0.1.8; http://www.ebi.ac.uk/zerbino/oases/) (Schulz et al., 2012). Adapter sequences, low-quality reads, and short sequences ( < 100 bp) were removed using a custom Perl Program (Stajich et al., 2002). The GetORF program in the EMBOSS suite was used to predict open reading frames (ORFs) in the assembled unigenes (Rice et al., 2000). The ORFs were searched using BLASTx against the NCBI nonredundant (NR) and Swiss-Prot protein database with an E-value threshold of le-3. Functional annotation was performed based on the BLASTp results against the Swiss Prot and TrEMBL databases using the GoPipe program based on the gene2go software (http://gopipe.fishgenome.org), to annotate the gene ontology (GO) terms (http://www.geneontology.org). Pathway annotations were assigned using Blastall to search against the Eukaryotic Orthologous Groups (KOG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (Kanehisa et al., 2010).
2.5 Analysis of differentially expressed genesFor analysis of gene expression, the number of expression tags in one library was calculated and normalized to fragments per kilo base of transcript per million mapped reads (FPKM) (Mortazavi et al., 2008). Then, the expression abundance of each gene between the control and Cd-treated groups was counted based on the MARS model (MA-plot-based method with Random Sampling model) (Wang et al., 2010). In our study, we used a stringent FDR (false discovery rate) value of 0.001 and the absolute value of |log2 FoldChange|≤1 as the thresholds to judge a significant difference in gene expression between groups. All differentially expressed genes were mapped to terms in the KEGG database for pathway enrichment analysis.
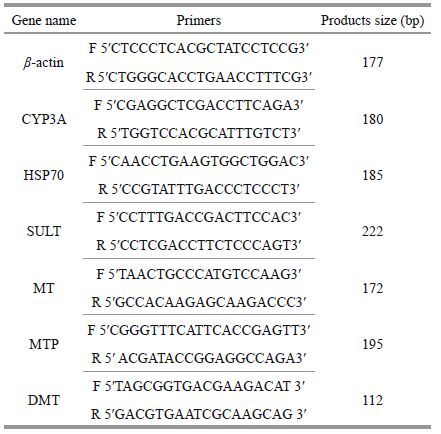
2.6 Quantitative real-time PCR (qRT-PCR) verificationSix differentially expressed genes were selected for validation by qRT-PCR using a SYBR Premix Ex Taq kit (Takara, Japan) according to the manufacturer's instructions. The qRT-PCRs were performed in triplicate on the Thermo System. Total RNA was isolated from the digestive glands of C. farreri using TRIzol reagent. β-Actin was used as an endogenous control. The specific primers used for the qRT-PCRs are listed in Table S1. The relative gene expression was analyzed using the comparative threshold cycle (CT) method established by Livak and Schmittgen (2001).
 |
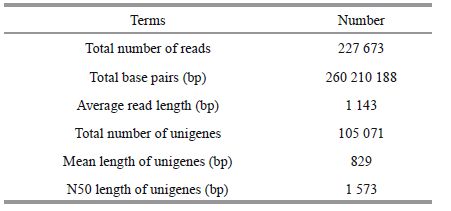
A cDNA pool was prepared from the digestive glands and sequenced to obtain a global overview of C. farreri transcriptome and related genes. We obtained approximately 227 673 transcripts. The raw reads produced have been deposited in the NCBI SRA database (accession number: SRR1563103). After adaptors and low-quality reads were removed, the filtered reads were de novo assembled with Trinity software. A total of 105 071 unigenes were obtained with an average size of 829 bp and a N50 length of 1 573 bp (Table 1). Among these unigenes, 61 828 (58.84%) were smaller than 500 bp, 19 393 (18.46%) were between 500 and 1 000 bp, 13 377 (12.73%) were between 1 000 and 2 000 bp, and 10 473 (9.97%) were longer than 2 000 bp. The length distribution of the unigenes is shown in Fig. 1.
 |

|
| Figure 1 Length distributions of clean reads The x-axis indicates sequence sizes (bp). The y-axis indicates the number of reads for every given size. |
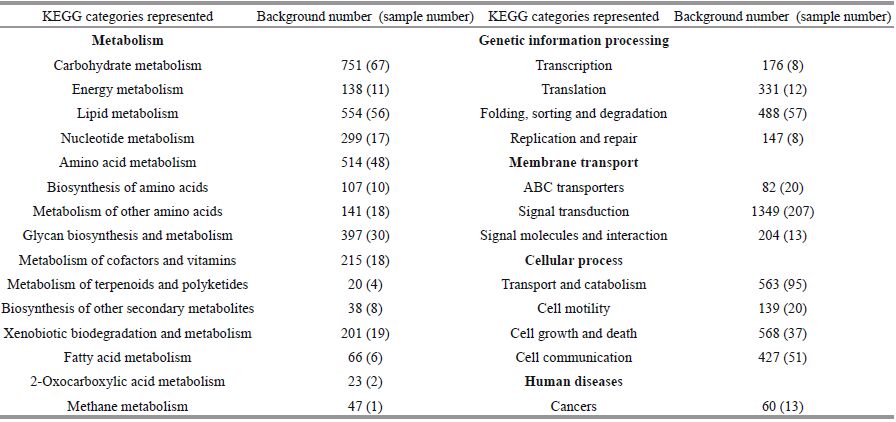
All the predicted protein sequences (unigene sequences were translated by the Transeq program of the EMBOSS suite) were searched against the NR and Swiss-Prot protein sequence databases for gene annotation. A total of 24 576 unigenes matched NR sequences and were assigned biological functions. Then, the functions were classified based on assigned GO terms. A total of 24 493 unigenes were assigned at least one GO term under one of the three categories: biological process, cellular component, and molecular function. Under biological process, cellular process (10.41%) and metabolic process (8.68%) were the most abundant terms; under molecular function, binding (9.81%) and catalytic activity (7.05%) were the most represented terms; and under cellular component, cell (5.41%) and cell part (5.40%) were the most represented terms (Fig. 2). These results are similar to the findings of a previous study on Mizuhopecten yessoensis exposed to Cd (Meng et al., 2013). Additionally, a total of 12 028 unigenes were classified functionally into 26 KOG families, including biochemical metabolism, signal transduction mechanism, defense system, cellular structure, and gene expression (Fig. 3). In total, 7849 unigenes were assigned into different KEGG pathways (Fig. 4). The most represented pathways were signal transduction (1 349 members), carbohydrate metabolism (751 members), and transport and catabolism (563 members) (Table 2). A wellcategorized and annotated resource of the C. farreri transcriptome could become a valuable database for investigating specific bioprocesses and identifying important functional genes in this important commercial species.

|
| Figure 2 GO annotations of transcriptomic data in C. farreri A total of 141 273 contigs are assigned to three GO terms including biological process, cellular component and molecular function. |

|
| Figure 3 KOG classification of the 105 071 unigenes Possible functions of unigenes were classified and subdivided into 26 categories. |

|
| Figure 4 KEGG classification of the unigenes Genes were divided into five branches according to the involvement of KEGG pathway. A: cellular processes; B: environmental information processing; C: genetic information processing; D: metabolism; E: organismal systems. |
Based on the GO annotation, we identified 24 493 sequences potentially related to response to a Cd stimulus that could be valuable for future research. Invertebrates have developed unique systems of biological host defense (so-called innate immunity) because they lack immunoglobulins (Iwanaga and Lee, 2005). However, the exact molecular basis of the immune system remains poorly understood. For example, some of the unigenes were annotated as encoding members of the HSP (heat shock protein) family, including HSP22, HSP70, HSP90, and HSP transcription factor, which is known to be involved in the stress response in many species (Gupta et al., 2010). The abundant expression of HSP family transcripts is consistent with a previous report (Hou et al., 2011). In addition, other unigenes that were predicted to encode proteins involved in biological detoxification processes were detected, such as the CYP family, glutathione S-transferases, glutathione peroxidase, catalase, and superoxide dismutase.
Although information of immune-related genes on bivalve has increased, the available knowledge is still fragmentary (Moreira et al., 2012). The KEGG analysis identified unigenes associated with immuneresponse pathways such as Toll-like receptor signaling pathway, chemokine signaling pathway, NOD-like receptor signaling pathway, and complement and coagulation cascades in the immune system. Toll-like receptors are an ancient family of pattern recognition receptors that play major roles in activating the immune system and detecting non-self substances.We also detected unigenes encoding C3 and C1q, which are central components in the complement system that consists of a tightly regulated network of proteins that play crucial roles in host defense and inflammation response. In addition, MAPK (Kaminska, 2005), PI3 K (Wymann and Marone, 2005), and p38 (Kumar et al., 2003) were identified in corresponding pathways that are crucial in cellular responses to external stress signals. Thus, we identified a large set of immune-and metabolicrelated genes in the transcriptome data, which may play important roles in C. farreri responses and may help significantly improve our understanding of the molecular mechanisms involved.
3.4 Function annotation of differentially expressed genesGenes that were differentially expressed by more than 2-fold between the control and Cd-treated groups, were mapped to terms in the KEGG database and compared with the transcriptome constructed from the pooled cDNA. A total of 3 800 differentially expressed sequences with KEGG annotations were observed between the Cd-treated and control groups (XC vs XD) (Fig. 5). Most of these genes were involved in biological processes such as ATP binding cassette (ABC) transporters, oxidative phosphorylation, ribosome biogenesis in eukaryotes, MAPK signaling pathway, glutathione metabolism, serine and threonine metabolism, and steroid hormone biosynthesis.

|
| Figure 5 Volcanic map of analysis of differentially expressed gene between samples The x-axis indicates the fold change of gene expression in different samples. The y-axis indicates the statistically significant degree of change in gene expression level. When corrected P-value is smaller, -log10 (corrected P-value) is bigger, which showing the more significant the difference of gene expression. The scatters represent genes. The blue dot indicates no significant difference, red one indicates significantly difference of up-regulated genes, green one indicates down-regulated genes significantly. |
Following Cd exposure, several protein families including the ABC transporter, HSP, and CYP families were found to be up-regulated in the digestive gland of C. farreri. Previous work has indicated that ABCBand ABCC-type transporters form an environmenttissue barrier in bivalve gills and confer multixenobiotic resistance (Luckenbach and Epel, 2008). This protective mechanism is supposedly ubiquitous in aquatic organisms. Additionally, another research has suggested that the ABC multidrug transporters play an important role in detoxifying mechanism of various toxic chemicals in aquatic invertebrates (Kingtong et al., 2007). These findings may explain why we found that the ABC families were significantly up-regulated in the digestive gland. CYP3A, a member of the cytochrome P450 family, is known to be involved in the metabolism of a wide range of drugs in many organisms, and it is strongly induced by a variety of structurally unrelated xenobiotics (Hegelund and Celander, 2003). In mammals, erythromycin-N-demethylase (ERND) was found to be partially linked to CYP3A, and is considered as a useful biomarker to evaluate the adverse effect of environmental pollutants (Vaccaro et al., 2003). Previous studies have demonstrated that the expression of CYP3A in some fish species could be induced by certain xenobiotics (Hasselberg et al., 2004; Meucci and Arukwe, 2006). However, in this study, CYP3A was down-regulated in the digestive gland, probably because of the different species, tissues, or exposure times, dosages, and xenobiotics that were used. Metallothionein (MT) is a cysteinerich, low-molecular-weight protein that plays an important role in metal metabolism and detoxification (Olsson et al., 1989). It was reported that MT could be used as a bioindicator of metal contamination in aquatic environments through binding to particular metals. In the digestive gland of the edible mussel Mytilus edulis exposed to 200 μg/L Cd for 4 days, Geret and Cosson (2002) observed a significant increase of MT protein levels compared with a control. As reported by Zorita et al. (2007) an increase on the content of MT in the digestive gland of mediterranean mussels (Mytilus galloprovincialis) was induced by 200 μg/L Cd after 2 and 9 days of exposure. The levels of MT were also induced by 34 μg/L Cd for 5 days and 20 μg/L Cd for 63 days on zebra mussel (Dreissena polymorpha) (Lecoeur et al., 2004; Faria et al., 2009). In this study, the expression of MT was induced by 0.2 mg/L Cd significantly. Thus, the synthesis of MT has been found to increase significantly in response to Cd exposure as a protection mechanism against Cd toxicity. Metal tolerance protein (MTP) belongs to the cation diffusion facilitator protein family, which is widespread in bacteria, fungi, plants, and animals. MTP is a bivalent cation transporter that is necessary for efficient translocation of Zn, Cd, and other heavy metals (Ricachenevsky et al., 2013). In this study, exposure to Cd induced the expression of MTP in the C. farreri digestive gland. The transporter for nonheme iron at the luminal intestinal surface is divalent metal transporter 1 (DMT1), which is an H+-coupled and electrogenic membrane transporter that belongs to a large family of integral membrane proteins highly conserved throughout evolution. In our study, the expression of DMT1 decreased in the Cd-treated group, which may indicate that Cd inhibited the transportation of iron, as reported previously (Bannon et al., 2003).
3.5 Experimental validation of differentially expressed genesSix differentially expressed genes were selected for validation by qRT-PCR. The results showed that five of the six primers produced bands of the expected sizes, and the five PCR products were further confirmed by Sanger sequencing (data not shown). However, all six genes showed the same expression change trends as the corresponding expressions estimated based on the transcriptomics data, which confirmed the effectiveness of our comparative transcriptomics analysis (Fig. 6).

|
| Figure 6 Quantitative RT-PCR validation of differentially expressed genes between control and Cd exposure groups CYP3A: cyto-chrome P450, family 3, subfamily A; HSP70: heat shock protein 70; SULT: sulfotransferase; MT: metallothionein; MTP: metal tolerance protein; DMT1: divalent metal transporter 1. Verticalbars represent the mean±SD (n=3) relative to the control. |
The results were replicated in a second experiment. The Pearson correlation was 0.852 for XC1 vs XC2, and 0.86 for XD1 vs XD2 (Fig. 7).

|
| Figure 7 The pearson correlation between samples The x-axis and y-axis represent log10 (FPKM+1) of different samples; R2: square of pearson correlation; rho: spearman correlation; tau: kendall-tau correlation. |
In this study, we generated a comprehensive transcriptome of adult C. farreri based on the Illumina sequencing platform. A total of 105 071 unigenes were assembled from the transcripts and 24 576 of them were assigned explicit annotation. A significant number of genes involved in the metabolic immune pathway, and response to a Cd stimulus were identified in the transcriptome. These findings substantially supplement existing genomic resources for C. farreri without reference genome information. In addition, differentially expressed genes were identified and functionally annotated based on database. The genetic information provided potential molecular markers in bivalves exposed to marine pollutants for functional studies of genes and better understanding of molecular mechanisms involved in metabolic and immune pathways.
5 ACKNOWLEDGMENTThe authors thank Dr. Yuefeng CAI (Ocean University of China) for useful advice and assistance.
| Bannon D I, Abounader R, Lees P S J, Bressler J P, 2003. Effect of DMT1 knockdown on iron, cadmium, and lead uptake in Caco-2 cells. Am. J. Physiol. Cell Physio., 284(1): C44–C50. Doi: 10.1152/ajpcell.00184.2002 |
| Chelomin V P, Bobkova E A, Lukyanova O N, Chekmasova N M, 1995. Cadmium-induced alterations in essential trace element homoeostasis in the tissues of scallop Mizuhopecten yessoensis. Comp. Biochem. Phys. C, 110(3): 329–335. |
| Faria M, Carrasco L, Diez S, Riva M C, Bayona J M, Barata C, 2009. Multi-biomarker responses in the freshwater mussel Dreissena polymorpha exposed to polychlorobiphenyls and metals. Comp. Biochem. Phys. C, 149(3): 281–288. |
| Garber M, Grabherr M G, Guttman M, Trapnell C, 2011. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods, 8(6): 469–477. Doi: 10.1038/nmeth.1613 |
| Gavery M R, Roberts S B, 2012. Characterizing short read sequencing for gene discovery and RNA-Seq analysis in Crassostrea gigas. Comp. Biochem. Phys. D, 7(2): 94–99. |
| Geret F, Cosson R P, 2002. Induction of specific isoforms of metallothionein in mussel tissues after exposure to cadmium or mercury. Arch. Environ. Contam. Toxicol., 42(1): 36–42. Doi: 10.1007/s002440010289 |
| Gupta S C, Sharma A, Mishra M, Mishra R K, Chowdhuri D K, 2010. Heat shock proteins in toxicology: how close and how far?. Life Sci., 86(11-12): 377–384. Doi: 10.1016/j.lfs.2009.12.015 |
| Hasselberg L, Meier S, Svardal A, Hegelund T, Celander M C, 2004. Effects of alkylphenols on CYP1A and CYP3A expression in first spawning Atlantic cod (Gadus morhua). Aquat. Toxicol., 67(4): 303–313. Doi: 10.1016/j.aquatox.2003.12.007 |
| Haynes D, Johnson J E, 2000. Organochlorine, heavy metal and polyaromatic hydrocarbon pollutant concentrations in the Great Barrier Reef (Australia) environment: a review. Mar. Pollut. Bull., 41(7-12): 267–278. Doi: 10.1016/S0025-326X(00)00134-X |
| He L, Wang Q, Jin X K, Wang Y, Chen L L, Liu L H, Wang Y, 2012. Transcriptome profiling of testis during sexual maturation stages in Eriocheir sinensis using Illumina sequencing. PLoS One, 7(3): e33735. Doi: 10.1371/journal.pone.0033735 |
| Hegelund T, Celander M C, 2003. Hepatic versus extrahepatic expression of CYP3A30 and CYP3A56 in adult killifish (Fundulus heteroclitus). Aquat. Toxicol., 64(3): 277–291. Doi: 10.1016/S0166-445X(03)00057-2 |
| Hou R, Bao Z M, Wang S, Su H L, Li Y, Du H X, Hu J J, Wang S, Hu X L, 2011. Transcriptome sequencing and De Novo analysis for yesso scallop (Patinopecten yessoensis) using 454 GS FLX. PLoS One, 6(6): e21560. Doi: 10.1371/journal.pone.0021560 |
| Hsu J C, Chien T Y, Hu C C, et al, 2012. Discovery of genes related to insecticide resistance in Bactrocera dorsalis by functional genomic analysis of a De Novo assembled transcriptome. PLoS One, 7(8): e40950. Doi: 10.1371/journal.pone.0040950 |
| Iwanaga S, Lee B L, 2005. Recent advances in the innate immunity of invertebrate animals. J. Biochem. Mol. Biol., 38(2): 128–150. |
| Kaminska B, 2005. MAPK signalling pathways as molecular targets for anti-inflammatory therapy-from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta, 1754(1-2): 253–262. Doi: 10.1016/j.bbapap.2005.08.017 |
| Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M, 2010. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res., 38(1): D355–D360. |
| Kingtong S, Chitramvong Y, Janvilisri T, 2007. ATP-binding cassette multidrug transporters in Indian-rock oyster Saccostrea forskali and their role in the export of an environmental organic pollutant tributyltin. Aquat. Toxicol., 85(2): 124–132. Doi: 10.1016/j.aquatox.2007.08.006 |
| Kumar S, Boehm J, Lee J C, 2003. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov., 2(9): 717–726. Doi: 10.1038/nrd1177 |
| Lecoeur S, Videmann B, Berny P, 2004. Evaluation of metallothionein as a biomarker of single and combined Cd/Cu exposure in Dreissena polymorpha. Environ. Res., 94(2): 184–191. Doi: 10.1016/S0013-9351(03)00069-0 |
| Livak K J, Schmittgen T D, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods, 25(4): 402–408. Doi: 10.1006/meth.2001.1262 |
| Luckenbach T, Epel D, 2008. ABCB-and ABCC-type transporters confer multixenobiotic resistance and form an environment-tissue barrier in bivalve gills. Am. J. Physiol. Regul. Integr. Comp. Physiol., 294(6): R1919–R1929. Doi: 10.1152/ajpregu.00563.2007 |
| Lukyanova O N, Khotimchenko Y S, 1995. Lipid peroxidation in organs of the scallop Mizuhopecten yessoensis and seaurchin Strongylocentrotus intermedius during the reproductive cycle. Comp. Biochem. Phys. B, 110(2): 371–377. Doi: 10.1016/0305-0491(94)00154-M |
| Meng X L, Liu M, Jiang K Y, Wang B J, Tian X, Sun S J, Luo Z Y, Qiu C W, Wang L, 2013. De Novo characterization of Japanese scallop Mizuhopecten yessoensis transcriptome and analysis of its gene expression following cadmium exposure. PLoS One, 8(5): e64485. Doi: 10.1371/journal.pone.0064485 |
| Meng X L, Tian X, Liu M, Nie G X, Jiang K Y, Wang B J, Wang L, 2014. The transcriptomic response to copper exposure by the gill tissue of Japanese scallops (Mizuhopecten yessoensis) using deep-sequencing technology. Fish Shellfish Immunol., 38(2): 287–293. Doi: 10.1016/j.fsi.2014.03.009 |
| Meucci V, Arukwe A, 2006. The xenoestrogen 4-nonylphenol modulates hepatic gene expression of pregnane X receptor, aryl hydrocarbon receptor, CYP3A and CYP1A1 in juvenile Atlantic salmon (Salmo salar). Comp. Biochem. Phys. C, 142(1-2): 142–150. |
| Moreira R, Balseiro P, Planas J V, Fuste B, Beltran S, Novoa B, Figueras A, 2012. Transcriptomics of In vitro immunestimulated hemocytes from the manila clam Ruditapes philippinarum using high-throughput sequencing. PloS One, 7(4): e35009. Doi: 10.1371/journal.pone.0035009 |
| Mortazavi A, Williams B A, McCue K, Schaeffer L, Wold B, 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods, 5(7): 621–628. Doi: 10.1038/nmeth.1226 |
| Nagashima K, Sato M, Kawamata K, Nakamura A, Ohta T, 2005. Genetic structure of Japanese scallop population in Hokkaido, analyzed by mitochondrial haplotype distribution. Mar. Biotechnol., 7(1): 1–10. Doi: 10.1007/s10126-004-3046-9 |
| Olsson P E, Larsson Å, Maage A, Haux C, Bonham K, Zafarullah M, Gedamu L, 1989. Induction of metallothionein synthesis in rainbow trout, Salmo gairdneri, during long-term exposure to waterborne cadmium. Fish Physiol. Biochem., 6(4): 221–229. Doi: 10.1007/BF01875025 |
| Ou J T, Meng Q G, Li Y, et al, 2012. Identification and comparative analysis of the Eriocheir sinensis microRNA transcriptome response to Spiroplasma eriocheiris infection using a deep sequencing approach. Fish Shellfish Immunol., 32(2): 345–352. Doi: 10.1016/j.fsi.2011.11.027 |
| Peng S T, Hu Y D, Bai Z P, 2009. Pollution assessment and ecological risk evalution for heavy metals in the sediments of Bohai Bay. J. Waterway Harb., 30(1): 57–60. |
| Ricachenevsky F K, Menguer P K, Sperotto R A, Williams L E, Fett J P, 2013. Roles of plant metal tolerance proteins (MTP) in metal storage and potential use in biofortification strategies. Front. Plant Sci., 4: 144. |
| Rice P, Longden I, Bleasby A, 2000. EMBOSS: the European molecular biology open software suite. Trends Genet., 16(6): 276–277. Doi: 10.1016/S0168-9525(00)02024-2 |
| Sato M, Kawamata K, Zaslavskaya N, et al, 2005. Development of microsatellite markers for Japanese scallop (Mizuhopecten yessoensis) and their application to a population genetic study. Mar. Biotechnol., 7(6): 713–728. Doi: 10.1007/s10126-004-0127-8 |
| Schulz M H, Zerbino D R, Vingron M, Birney E, 2012. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 28(8): 1086–1092. Doi: 10.1093/bioinformatics/bts094 |
| Shan Z G, Li H J, Bao X B, et al, 2011. A selenium-dependent glutathione peroxidase in the Japanese scallop, Mizuhopecten yessoensis: cDNA cloning, promoter sequence analysis and mRNA expression. Comp. Biochem. Phys. B, 159(1): 1–9. Doi: 10.1016/j.cbpb.2011.01.003 |
| Slobodskova V V, Zhukovskaya A F, Chelomin V P, 2012. DNA damage in the gill cells of the marine scallop Mizuhopecten yessoensis during anoxic stress and aerobic recovery. Ocean Sci. J., 47(2): 95–100. Doi: 10.1007/s12601-012-0010-x |
| Stajich J E, Block D, Boulez K, et al, 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res., 12(10): 1611–1618. Doi: 10.1101/gr.361602 |
| Torres M A, Barros M P, Campos S C G, Pinto E, Rajamani S, Sayre R T, Colepicolo P, 2008. Biochemical biomarkers in algae and marine pollution: a review. Ecotoxicol. Environ. Saf., 71(1): 1–15. Doi: 10.1016/j.ecoenv.2008.05.009 |
| Vaccaro E, Giorgi M, Longo V, Mengozzi G, Gervasi P G, 2003. Inhibition of cytochrome p450 enzymes by enrofloxacin in the sea bass (Dicentrarchus labrax). Aquat. Toxicol., 62(1): 27–33. Doi: 10.1016/S0166-445X(02)00064-4 |
| Wang L K, Feng Z X, Wang X, Wang X W, Zhang X G, 2010. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics, 26(1): 136–138. Doi: 10.1093/bioinformatics/btp612 |
| Wang X J, Hu X L, Li J Q, et al, 2012. Characterization of 38 EST-derived SNP markers in Zhikong scallop (Chlamys farreri) and their cross-species utility in Yesso scallop (Patinopecten yessoensis). Conserv. Genet. Resour., 4(3): 747–753. Doi: 10.1007/s12686-012-9636-3 |
| Wymann M P, Marone R, 2005. Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr. Opin. Cell Biol., 17(2): 141–149. Doi: 10.1016/j.ceb.2005.02.011 |
| Zerbino D R, Birney E, 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res., 18(5): 821–829. Doi: 10.1101/gr.074492.107 |
| Zhao X X, Wang Q H, Jiao Y, Huang R L, Deng Y W, Wang H, Du X D, 2012. Identification of genes potentially related to biomineralization and immunity by transcriptome analysis of Pearl Sac in Pearl Oyster Pinctada martensii. Mar. Biotechnol., 14(6): 730–739. Doi: 10.1007/s10126-012-9438-3 |
| Zorita I, Bilbao E, Schad A, Cancio I, Soto M, Cajaraville M P, 2007. Tissue-and cell-specific expression of metallothionein genes in cadmium-and copper-exposed mussels analyzed by in situ hybridization and RT-PCR. Toxicol. Appl. Pharmcol., 220(2): 186–196. Doi: 10.1016/j.taap.2007.01.003 |