 2018, Vol. 36
2018, Vol. 36Institute of Oceanology, Chinese Academy of Sciences
Article Information
- ZHANG Yaoling(张耀玲), YANG Keli(杨克利), DU Jinzhou(杜金洲), ZHANG Fenfen(张芬芬), DONG Yaping(董亚萍), LI Wu(李武)
- Chemical characterization of fractions of dissolved humic substances from a marginal sea-a case from the Southern Yellow Sea
- Chinese Journal of Oceanology and Limnology, 36(2): 238-248
- http://dx.doi.org/10.1007/s00343-017-6202-6
Article History
- Received Aug. 2, 2016
- accepted in principle Sep. 22, 2016
- accepted for publication Nov. 17, 2016




2 State Key Laboratory of Estuarine and Coastal Research, East China Normal University, Shanghai 200062, China
Marine dissolved organic carbon (DOC) represents one of the largest dynamic pools of organic carbon in the global carbon cycle, and it is approximately equal to the amount of organic carbon in the terrestrial biosphere and the amount of carbon in the atmosphere as CO2 (Hedges, 1992; Hedges et al., 1997). Even a very small change of the marine DOC pool can potentially impact many of the earth's biogeochemical processes. For example, oxidation of just 1% of the marine DOC pool would generate a CO2 flux larger than that annually produced by fossil fuel combustion (Mopper et al., 2007). Despite of the importance of marine dissolved organic matter (DOM), only a small fraction of DOM has been identified, including carbohydrates, amino acids, lipids, and amino sugars (Koch et al., 2005), hindering our understanding of DOM cycling and sources in the marine environment. The characterization of marine DOM has always been constrained by the inability to obtain a representative fraction of the DOM pool for analysis, and its intrinsic chemical complexity (Simjouw et al., 2005; Mao et al., 2012). Nevertheless, by using advanced instrumental approaches recently, such as Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) and advanced one-and two-dimensional nuclear magnetic resonance (NMR) techniques, some new classes of compounds were reported for marine DOM. For example, carboxyl-rich alicyclic molecules (CRAM) were reported to be an important component of marine and freshwater DOM (Hertkorn et al., 2006; Lam et al., 2007; Cao et al., 2016). The material derived from linear terpenoids (MDLT) was also reported to contribute significantly to natural water DOM (Lam et al., 2007; Woods et al., 2010).
Generally, three strategies are employed to isolate DOM from seawaters. They are solid-phase extractions (SPE) with XAD resins, C18, or PPL adsorbent under acidic conditions; tangential-flow ultrafiltration (UF); and reverse osmosis/ electrodialysis (RO/ED) (Perdue and Benner, 2009; Nebbioso and Piccolo, 2013). Higher DOC recoveries, approximately 82% for deep water and 75% for surface water, were achieved by the RO/ED method (Koprivnjak et al., 2009; Green et al., 2014). Relatively lower DOC recoveries were acquired for SPE and UF methods, with 17%–61% for SPE and 13%–38% for UF, respectively (Perdue and Benner, 2009; Green et al., 2014). Different properties were also reported for marine DOM isolated by different methods. For example, SPE samples are typically enriched in chromophores, fluorophores, and aromatic compounds; UF samples contain a relatively larger high-molecular-weight fraction of marine DOM; RO/ ED samples present the combined features of UF and SPE samples, with different acid-base properties than SPE samples (Koprivnjak et al., 2009; Perdue and Benner, 2009; Zhang et al., 2013b). So far, there is no single method that can achieve quantitative isolation of all organic components from an aquatic environment (Leenheer, 1981). Due to the inherent complexity and heterogeneity of marine DOM, it is difficult, in general, to acquire the composite, structural and functional features of DOM with a single technique. The combination of multiple analytical methods is a more useful strategy for studying such complex material. 13C cross polarization magic angle spinning (CP/MAS) NMR technique providing a useful analytical window into DOM composition with the benefit of being non-destructive has been routinely used to analyze the structural information of marine DOM in many studies (Benner et al., 1992; Hedges et al., 1992; Esteves et al., 2009; Koprivnjak et al., 2009). Based on different electronic environments of specific nuclei in DOM, NMR technique has the ability to evaluate the distribution of DOM functional groups (Abdulla et al., 2010; Tremblay et al., 2011). On the basis of the vibration of molecules, Fourier transform infrared spectroscopy (FTIR) technique can investigate additional information of the functional groups in DOM, such as resolving the carboxyl, amide and aliphatic ester contributions (Abdulla et al., 2010). Pyrolysis gas chromatography/ mass spectrometry (Py-GC/MS) is a powerful tool for studying complex DOM. It allows the identification of individual functional groups and structural moieties after heat-induced breakdown to smaller units (Kisand et al., 2013).
Due to the wide use and relatively low cost of XAD resins (Green et al., 2014), XAD-8 and XAD-4 resins were used to isolate marine DOM from coastal water in the Southern Yellow Sea (SYS), China. XAD methods were originally developed to isolate operationally defined "humic" fractions of DOM (Aiken, 1985; Aiken et al., 1992), and thus the isolated marine DOM is subdivided into three dissolved humic fractions. Multiple analytical techniques were used to characterize the isolated DOM to better understand its origin, composition, and cycling in coastal areas from the marginal sea including stable carbon isotope analysis, FTIR, 13C CP/MAS NMR, and Py-GC/MS analyses.
2 MATERIAL AND METHOD 2.1 Sampling sites descriptionThe Yellow Sea, which is bordered by the Chinese and Korean Peninsulas, is a typical epicontinental sea of the northwest Pacific Ocean. The region of the SYS is a highly biologically active area with complicated hydrologic variations. This area is influenced by strong tidal current and freshwater discharges, such as the Yellow Sea Cold Water Mass, the Yellow Sea Warm Current, and the Yellow Sea Coastal Current, etc.; and the mean and maximum depths are 44 and 103 m, respectively (Jacobs et al., 2000; Bai et al., 2014). Two seawater samples were collected from surface water (depth=2 m) by using a peristaltic pump during an August 2009 research cruise in the SYS aboard the R/V Beidou. The sampling sites are shown in Fig. 1. Approximately 1 000 L of seawater were collected in pre-cleaned containers from the 10494 site (water depth 51 m) and the 82194 site (water depth 80 m), respectively. Immediately following collection the seawater samples were sequentially filtered through 1.0 μm and 0.45 μm filters. Then, the filtered samples were kept in the dark for further dissolved humic substances (DHS) extractions in the field.

|
| Figure 1 The sampling locations of the two seawater samples |
The methods used for marine DHS isolation were provided by Aiken (1985) and Aiken et al. (1992). Briefly, the filtered samples were acidified to pH 2.0 with HCl and passed through an XAD-8 and XAD-4 resin (Amberlite, USA) in series. The procedures for resin cleaning were according to Thurman and Malcolm (1981). Each XAD resin column was backeluted separately with 0.1 mol/L NaOH and the eluates were acidified immediately. The eluate from XAD-8 resin was re-concentrated with a smaller XAD-8 column, and the NaOH eluate was acidified to pH 1.0 with HCl. Then, the humic acids (HAs) were separated from the fulvic acids (FAs) by centrifuge. The HAs were dissolved by adding sufficient 0.1 mol/L NaOH and then acidified by passing through an H+-saturated cation exchange resin. The FAs were purified by readsorption on XAD-8 resin. The alkaline eluate from the XAD-8 resin was protonated by using an H+-saturated cation exchange resin. The eluate from the XAD-4 resin was also re-adsorbed on a smaller XAD-4 resin column, and the alkaline eluate was protonated by using an H+-saturated cation exchange resin. The FAs, HAs and the XAD-4 fractions were freeze-dried for following analysis.
2.3 Chemical characterization of marine DHS 2.3.1 Stable isotopic compositionThe stable carbon isotope 13C was analyzed in duplicate with a Delta plus XP isotope ratio mass spectrometer (Thermo Finnigan, USA). The results were reported in standard delta notation relative to the conventional Pee Dee Belemnite (13C/12C, PDB) standards.
2.3.2 Fourier transform infrared spectroscopyEach FA, HA and the XAD-4 fraction was prepared as KBr discs where a 1.0 mg sample was diluted with 100.0 mg KBr. FTIR spectra were obtained by collecting 200 scans with Nicolet 8700 USB FTIR spectrometer. Spectra were acquired from 4 000 to 400 cm-1 at 4 cm-1 resolution.
2.3.3 Solid-state 13C nuclear magnetic resonanceAll solid-state 13C NMR analyses were performed at 100 MHz using a Bruker Avance DSX 300 MHz spectrometer. Spectra were acquired by using the CP/ MAS technique. Each sample was packed into a zirconia rotor and spun at 5 kHz with a pulse delay of 1 s during data acquisition. Qualitative composition information was obtained by 13C CP/TOSS (crosspolarization/total sideband suppression) NMR. The contact time was 1 ms and chemical shifts were externally referenced to the glycine resonance at 176×10-6.
2.3.4 Pyrolysis gas chromatography mass spectrometryPyrolysis-GC/MS was performed on a QP2010 gas chromatography mass spectrometer (Shimadzu, Japan) operating in split mode, equipped with a Chemical Data System 2000 (CDS Analytical Inc., USA) pyrolysis interface. The GC instrument was equipped with an HP-5 MS capillary column (30 m×0.25 mm i.d., 0.25 μm film thickness). Approximately 0.5 mg samples were heated from 250 to 610℃ at a rate of 5℃/ms and held for 10 s. The split ratio and inlet temperature were 1:40 and 250℃, respectively. The initial oven temperature was 40℃ (2 min), then heated at 8℃ /min to 310℃, and maintained at that temperature for 10 min. The flow rate is 1 mL/min with helium as a carrier gas. A mass detection range of m/z 45–650 was obtained using mass spectra at 70 eV ionization energy. The compound identifications were according to the NIST library, GC retention times, and other publications (Chefetz et al., 2000; Buurman et al., 2009; de la Rosa et al., 2011; Zhang et al., 2016). The sum of the peak areas of the total identified pyrolysis compounds was taken as 100%. The relative abundance of each pyrolysis product was calculated on this basis using the total ion current (TIC) programs.
3 RESULT AND DISCUSSIONThe DOC concentrations for the surface water in the studied areas were reported approximately in the range of 1.4 to 2.0 mg/L in April 2007 (Xie et al., 2010), 1.5 to 1.8 mg/L in September 2010 (Li et al., 2013), and 1.7 to 2.1 mg/L in August 2013 (Yuan et al., 2015), respectively. The DOC recoveries in this study were not determined; however, XAD resins were reported to recover 35%–42% of DOC from seawater in general, with 25% from XAD-8 resin column and 10%–17% from XAD-4 resin column (Esteves et al., 2009; Green et al., 2014). Among the final freeze-dried samples, FAs and the XAD-4 fractions were dominant products while only a few milligrams of HAs were obtained, and this phenomenon was also observed in other studies (Esteves et al., 2007, 2009; Zhang et al., 2013a).
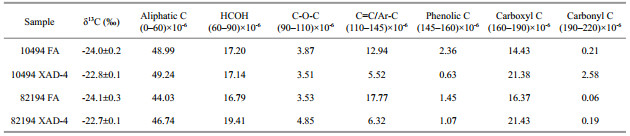
3.1 Stable carbon isotopic compositionIt is reported that typical δ13C values for marine algae and plankton usually fell between -22‰ and -19‰, while most terrestrial plants which use the C3 Calvin pathway to combine carbon into organic matter have average δ13C values of ~-27‰ (Meyers, 1994; Hedges et al., 1997). The δ13C values for FAs and the XAD-4 fractions are listed in Table 1. Due to the limited quantity of HA fractions, their δ13C values were not determined. The δ13C values for the two DHS samples ranged from -24.1‰ to -22.7‰, with relatively higher values for the XAD-4 fractions than for FA samples. The δ13C values for the DHS from the two sites were very similar. It can be inferred that the DHS of the two sites in this study mainly came from a marine source with minor contributions from terrestrial materials. Due to the semi-enclosed water body system of the SYS, the terrestrial inputs from rivers and tidal currents may influence the sampling areas. The XAD-4 fractions have obviously higher δ13C values than the FAs, which agrees well with other studies (Esteves et al., 2007; Zhang et al., 2013a). Esteves et al. (2007) provided two hypotheses for the formation mechanism of the XAD-4 fraction: one assumption is the XAD-4 fraction consists of an older resistant fraction of marine DHS, another assumes the XAD-4 fraction is the most labile and young part of the DHS. Each of these hypotheses led to a different pathway for the formation of the XAD-4 fraction.
|
The FTIR spectra of the marine DHS are shown in Fig. 2. The spectra of the samples in this study generally exhibited similar features due to overlapping absorption from these heterogeneous mixtures. The common broad band approximately 3 400 cm-1 was attributed to the intra-and inter-molecular hydrogenbonded OH groups in phenols, carbohydrates and carboxylic acids compounds. Moreover, N-H groups also exhibited absorbance in this region, but it was generally centered at lower wavenumbers near 3 200 cm-1 (Stevenson, 1994; Abdulla et al., 2010; Tremblay et al., 2011). The peaks at 2 920 cm-1 and 2 850 cm-1 were ascribed to aliphatic CH2 stretching (Stevenson, 1994). The peak intensities decreased from HAs to FAs then to the XAD-4 fractions in both sites, which indicated that HAs contain more aliphatic moieties than the other two counterparts. The findings agreed well with our previous study for estuarine and coastal DHS (Zhang et al., 2013a).

|
| Figure 2 The FTIR spectra for the three fractions of the two DHS samples |
The peaks at 1 720 cm-1 and 1 220 cm-1 that are attributed to C=O and C-O stretching and O-H deformation of the protonated carboxylic acid functional groups (Stevenson, 1994), were very distinct for FAs and the XAD-4 fractions, but appeared only as a shoulder in HAs from the two sites. The adsorption approximately 1 400 cm-1, which was attributed to de-protonated symmetric stretching of carboxyl groups (Abdulla et al., 2010; Zhang et al., 2011), was also following the same variation trend. Both pieces of evidence indicate that HAs contain much less carboxyl groups than FAs and the XAD-4 fractions. The peak at 1 650 cm-1, which was attributed to C=O stretching in Amide I (Abdulla et al., 2010; Tremblay et al., 2011), was very obvious for HAs, but almost absent in FAs and the XAD-4 fractions. This phenomenon demonstrates that HAs contain more nitrogen-containing compounds than the other two counterparts.
The bands approximately 1 515 cm-1 were assigned to the aromatic C=C stretching and the C-O asymmetric stretching of phenol compounds (Esteves et al., 2009; Abdulla et al., 2010). The adsorptions at this band were obvious for HAs and FAs but absent for the XAD-4 fractions from the two sites. This phenomenon implied that, among the three fractions of DHS, HAs and FAs presented more aromatic features than the XAD-4 fractions. The adsorptions between 1 400–1 260 cm-1and 1 200–1 000 cm-1 were attributed to the vibration of C-O bands in carbohydrates (Abdulla et al., 2010). The intensities of these bands decreased slightly from the XAD-4 fractions to FAs and HAs in this study. Although the δ13C values were not determined for HAs, their higher values for the XAD-4 fractions may be attributed to the lower content of aromatics and higher percentage of carbohydrates because lignin compounds are depleted in 13C compared to the 13C-enriched carbohydrates and proteins (Hatcher et al., 1983; Benner et al., 1987).
3.3 13C CP/MAS NMR spectroscopyThe CP/MAS 13C NMR spectra and the percentage of the integrated peaks under each region of the spectra for the two DHS samples are shown in Fig. 3 and Table 1, respectively. Again, due to the small quantity of HA samples, their 13C NMR spectra were not determined. All spectra could be divided into seven chemical-shift regions: (0–60)×10-6, attributed to alkyl carbons; (60–90)×10-6, attributed to carbohydrate-like carbons; (90–110)×10-6, assigned to anomeric carbons; (110–145)×10-6, ascribed to aromatic carbon; (145–160)×10-6, attributed to oxygen bounded aromatic carbons; (160–190)×10-6, ascribed to COO and N-C=O; and (190–220)×10-6, attributed to ketone, quinine, or aldehyde carbon (Hedges et al., 1992; Mao et al., 2007, 2010, 2012; Esteves et al., 2009; Zhang et al., 2014).

|
| Figure 3 The 13C CP/MAS NMR spectra of the FAs and the XAD-4 fractions for the two DHS samples |
As shown in Table 1, the largest percentage of carbon in all the samples is aliphatic carbons, which accounted for 44.03% to 49.24% of the total carbons. The highest content of aliphatic components was consistent with previous reports for marine and river DHS (Hedges et al., 1992; Esteves et al., 2009; Zhang et al., 2013a; Cao et al., 2016), which indicated a predominantly autochthonous algal and microbially derived source. Moreover, the high content of aliphatic carbons may also indicate a high content of CRAM, as CRAM has been found to be an important component of both marine and freshwater DOM (Hertkorn et al., 2006; Lam et al., 2007; Cao et al., 2016). The second largest percentage of carbon is O-alkyl C, which includes carbohydrate-like carbons ((60–90)×10-6) and anomeric carbons ((90–110)×10-6) that are substructures of carbohydrates. The carbon percentage of these two parts was 20.32% to 24.26% of the total carbons, with the XAD-4 fractions slightly higher than or comparable to that of the FAs for the two sites. Cao et al. (2016) also indicated that the DOM fractions isolated by XAD-4 resin contained more oxygen-and nitrogen-containing organic compounds than that isolated by XAD-8 resin for a river DOM sample. The total content of aromatic ((110–145)×10-6) and phenolic carbons ((145– 160)×10-6) ranged from 6.15% to 19.22% for FAs and the XAD-4 fractions for both sites, with the content much lower for the XAD-4 fractions than for the FAs. These findings are consistent with the FTIR results. The minor resonances corresponding to phenolic carbons (< 3 area %) indicated that lignin structural units were limited and reinforced the hypothesis that primarily contributions from a marine source are in our samples as presented in the stable carbon isotopic composition and FTIR analysis. Carboxyl carbons accounted for between 14.43% to 21.43% of the total carbons in our samples. Similar results were also found for other marine DHS samples (Hedges et al., 1992; Esteves et al., 2009). Benner et al. (1992) indicated that marine DHS were composed primarily of the older and more refractory components of DOM and enriched in unsubstituted alkyl and carboxyl carbons. Marine DOM samples isolated by the RO/ ED method contained less carboxyl carbons than DHS samples (Zhang et al., 2013b). For both sites, the XAD-4 fractions contained more carboxyl and carbonyl carbons than FAs, which agreed well with previous studies for seawater and freshwater DHS (Aiken et al., 1992; Hedges et al., 1992; Esteves et al., 2009). Aiken et al. (1992) indicated that the sorption characteristics of XAD resins are mainly dependent on chemical composition, resin surface area and resin pore size. The XAD-4 resins possess greater surface area than XAD-8 resins and, therefore, have greater capacity for organic components with lower molecular weight and higher content of heteroatom and carboxyl groups. Those features of the XAD-4 fractions may give them considerable geochemical significance by playing important roles in such processes as trace metal binding, mineral weathering and water acidification in the marine environments (Aiken et al., 1992).
Comparing the DHS samples from the two sites, the 10494 site contained slightly higher aliphatic carbons but lower aromatic and carboxyl carbons than that of the 82194 site. Since the δ13C values for each fraction of the two samples of DHS were very similar, the small differences between carbons in the two sites may indicate the 82194 site encountered slightly more contributions from terrestrial sources that may not be reflected in the δ13C values.
3.4 Pyrolysis-Gas Chromatography/Mass Spectrometry analysisThe TIC programs for the Py-GC/MS analysis of the DHS samples and the major identified compounds in the chromatograms are shown in Fig. 4 and Table 2, respectively. The Py-GC/MS analysis detected 78 pyrolysis products. As shown in Table 2, most of the pyrolysis products were aliphatic compounds (A), other aromatic components (Ar), and polysaccharides (Ps). The relative content of nitrogen-containing compounds (N), nitriles and amides (Nit), phenols (Ph), polyaromatic components (Par), fatty acids (FA), and sulfur-containing compounds (S) was much lower.

|
| Figure 4 Py-GC/MS total ion current pyrogram for the three fractions of the two DHS samples Peak numbers refer to identified compounds as in Table 2. |

The aliphatic pyrolysis products were mainly comprised of C7–C17 straight-chain n-alkane and n-alkene doublets with a small amount of other alkanes and alkenes. Long-chain aliphatic compounds were mainly derived from lipids. In both sites the HA samples contained much more aliphatic components than the other two counterparts, which agreed well with the FTIR results. Kisand et al. (2013) also indicated by Py-GC/MS analysis that the HA fractions were characterized by more aliphatic lipids for the riverine DOM that had been degraded by estuarine and marine bacterial communities under a different salinity gradient.
Aromatic pyrolysis products can be subdivided into three groups, i.e., phenols, polyaromatic components, and other aromatic components. Other aromatic components were much more abundant than phenols and polyaromatic components in the studied samples. The other aromatic components mainly include benzene, toluene, styrene, and a series of C2– C4-alkylbenzenes. Benzene is generally attributed to a pyrolysis product of (poly) phenols, while toluene can be attributed to the pyrolysis product of either (poly) phenols or proteins. Moreover, benzene and toluene may also derive from black carbon (Buurman et al., 2009). Styrene is known to derive either from degraded lignin, tannins, and anthropogenic aromatic residues or from thermally altered organic matter residues (de la Rosa et al., 2011). It is reported the alkylbenzenes series may derive from the degradation of charred material or be the cyclization products of decarboxylated fatty acids (Buurman et al., 2009). Phenol and alkyl phenols are major phenolic compounds that have multiple origins such as being pyrolysates of lignin-containing materials, algal and microbial polyphenols, amino acids, cellulose, or polycarboxylic acids (Chefetz et al., 2000; de la Rosa et al., 2011). The polyaromatic components were mainly the derivates of naphthalene, and their contribution was minor. Lignin derived compounds were not detected in this study. Among the three fractions of DHS, FAs contain more aromatic pyrolysis products than the XAD-4 fractions and HAs.
The polysaccharide-derived compounds detected by Py-GC/MS for the studied samples included acetic acid, the derivates of furan and cyclopentene, cyclopentenones, levoglucosenone, limonene, and 2-coumaranone. Among those components, acetic acid had the largest contribution for the XAD-4 fractions and FAs. The total content of polysaccharidederived compounds had no obvious variation trend for the three fractions of DHS.
The protein/nitrogen-containing compounds in the studied DHS samples can be subdivided into heterocyclic N components and nitriles. The heterocyclic N compounds included pyridine, pyrrole and its derivatives. The nitriles were mainly 2-methyl butanenitrile, 4-methyl pentanenitrile, benzonitrile, and benzyl nitrile. The content of nitrogen-containing pyrolysate was higher in HAs than in the XAD-4 fractions and FAs, in line with the FTIR results and the previous research on riverine, estuarine, and marine DHS (Esteves et al., 2007; Zhang et al., 2013a). This phenomenon may indicate that there were more proteinaceous materials in HAs than in the other two counterparts for aquatic DHS.
A small amount of sulfur-containing compounds was also identified including sulfur dioxide and dimethyl disulfide. There was no obvious trend for the content of sulfur-containing compounds among the three fractions of the two DHS samples. Moreover, short-chain fatty acids such as 2-propenoic acid and hexanoic acid were also detected. The content of fatty acids was relatively higher for the XAD-4 fractions and FAs than the HAs. However, the relative abundances of fatty acids in the Py-GC/MS analysis were much lower than the results from 13C NMR analysis. The limited detection of fatty acids should be attributed to the incompatibility of free fatty acids with the apolar columns used in GC (Buurman et al., 2009; de la Rosa et al., 2011).
3.5 The implications of DHS in the Southern Yellow SeaAs important fraction of marine DOM, DHS play key roles in marine biogeochemical processes. In addition to being an important source of nutrients for marine microbes, marine DHS also play important roles in trace metal and organic pollutant chelation in marine environments, thereby influencing trace metal and organic toxicity and bioavailability (Mopper et al., 2007). In this study, different properties of the three fractions of DHS may reflect different degradation states and cycling processes for the marine DOM in the SYS. HAs only accounted for a very small fraction of the marine DHS. Esteves et al.(2007) indicated that HAs were the condensed products from FAs preserving the amino-acids from those condensation reactions in marine environments. FAs and the XAD-4 fractions were the primary contributors to the marine DHS samples. The FAs contained more aromatic components in this study and were considered as metastable molecules that characterize a transient state of diverse precursor compounds during their oxidation (Reemtsma et al., 2006). The XAD-4 fractions were enriched in 13C and carbohydrates and may be easier utilized by microbes. In addition, the lower molecular weight and higher content of heteroatom and carboxyl groups for the XAD-4 fractions may give them considerable geochemical significance, such as in the processes of trace metal binding, mineral weathering and water acidification in marine environments.
4 CONCLUSIONIn this study, two DHS samples that were isolated from coastal water in the SYS were well characterized using stable carbon isotopic composition, FTIR, 13C NMR, and Py-GC/MS. Based on the complementary results obtained from those multiple analysis, several conclusions can be drawn as follows:
1) The two coastal DHS samples presented similar properties and both of them were mainly influenced by marine sources with minor contributions from terrestrial materials. The DHS samples were enriched in 13C, aliphatic components, and carboxyl carbons but depleted in unsaturated compounds. The lignin derived pyrolysis products were very limited. Those features indicated a predominance of algal and microbial-derived precursors for the studied samples.
2) The three fractions of DHS showed some different properties for the two sites. FAs presented more aromatic features than HAs and the XAD-4 fractions. HAs contained more aliphatic lipids and proteinaceous materials than FAs and the XAD-4 fractions. The XAD-4 fractions were enriched in 13C and contain more carbohydrates, but less aromatic compounds than the other two counterparts. Moreover, the lower molecular weight and higher content of heteroatom and carboxyl groups for the XAD-4 fractions may provide new insight into geochemical processes in marine environments, such as in the aspects of trace metal species, bioavailability of pollutants, mineral weathering and water acidification.
| Abdulla H A N, Minor E C, Dias R F, Hatcher P G, 2010. Changes in the compound classes of dissolved organic matter along an estuarine transect:a study using FTIR and 13C NMR. Geochim. Cosmochim. Acta, 74(13): 3 815–3 838. Doi: 10.1016/j.gca.2010.04.006 |
| Aiken G R, McKnight D M, Thorn K A, Thurman E M, 1992. Isolation of hydrophilic organic acids from water using nonionic macroporous resins. Org. Geochem., 18(4): 567–573. Doi: 10.1016/0146-6380(92)90119-I |
| Aiken G R. 1985. Isolation and concentration techniques for aquatic humic substances. In: Aiken G R, McKnight D M, Wershaw R L, McCarthy P eds. Humic Substances in Soil, Sediment, and Water: Geochemistry, Isolation, and Characterization. John Wiley & Sons, New York. p. 363-386. |
| Bai Y, Su R G, Shi X Y, 2014. Assessing the dynamics of chromophoric dissolved organic matter in the southern Yellow Sea by excitation-emission matrix fluorescence and parallel factor analysis (EEM-PARAFAC). Cont. Shelf Res., 88: 103–116. Doi: 10.1016/j.csr.2014.07.011 |
| Benner R, Fogel M L, Sprague E K, Hodson R E, 1987. Depletion of 13C in lignin and its implications for stable carbon isotope studies. Nature, 329(6141): 708–710. Doi: 10.1038/329708a0 |
| Benner R, Pakulski J D, McCarthy M, Hedges J I, Hatcher P G, 1992. Bulk chemical characteristics of dissolved organic matter in the ocean. Science, 255(5051): 1 561–1 564. Doi: 10.1126/science.255.5051.1561 |
| Buurman P, Nierop K G J, Kaal J, Senesi N, 2009. Analytical pyrolysis and thermally assisted hydrolysis and methylation of EUROSOIL humic acid samples-a key to their source. Geoderma, 150(1-2): 10–22. Doi: 10.1016/j.geoderma.2008.12.012 |
| Cao X Y, Aiken G R, Spencer R G M, Butler K, Mao J D, Schmidt-Rohr K, 2016. Novel insights from NMR spectroscopy into seasonal changes in the composition of dissolved organic matter exported to the Bering Sea by the Yukon River. Geochim. Cosmochim. Acta, 181: 72–88. Doi: 10.1016/j.gca.2016.02.029 |
| Chefetz B, Chen Y, Clapp C E, Hatcher P G, 2000. Characterization of organic matter in soils by thermochemolysis using tetramethylammonium hydroxide (TMAH). Soil Sci. Soc. Am. J., 64(2): 583–589. Doi: 10.2136/sssaj2000.642583x |
| de la Rosa J M, González-Pérez J A, González-Vila F J, Knicker H, Araújo M F, 2011. Molecular composition of sedimentary humic acids from South West Iberian Peninsula:a multi-proxy approach. Org. Geochem., 42(7): 791–802. Doi: 10.1016/j.orggeochem.2011.05.004 |
| Esteves V I, Otero M, Duarte A C, 2009. Comparative characterization of humic substances from the open ocean, estuarine water and fresh water. Org. Geochem., 40(9): 942–950. Doi: 10.1016/j.orggeochem.2009.06.006 |
| Esteves V I, Otero M, Santos E B H, Duarte A C, 2007. Stable carbon isotope ratios of tandem fractionated humic substances from different water bodies. Org. Geochem., 38(6): 957–966. Doi: 10.1016/j.orggeochem.2007.02.006 |
| Green N W, Perdue E M, Aiken G R, Butler K D, Chen H M, Dittmar T, Niggemann J, Stubbins A, 2014. An intercomparison of three methods for the large-scale isolation of oceanic dissolved organic matter. Mar. Chem., 161: 14–19. Doi: 10.1016/j.marchem.2014.01.012 |
| Hatcher P G, Spiker E C, Szeverenyi N M, Maciel G E, 1983. Selective preservation and origin of petroleum-forming aquatic kerogen. Nature, 305(5934): 498–501. Doi: 10.1038/305498a0 |
| Hedges J I, Hatcher P G, Ertel J R, Meyers-Schulte K J, 1992. A comparison of dissolved humic substances from seawater with Amazon River counterparts by 13C-NMR spectrometry. Geochim. Cosmochim. Acta, 56(4): 1 753–1 757. Doi: 10.1016/0016-7037(92)90241-A |
| Hedges J I, Keil R G, Benner R, 1997. What happens to terrestrial organic matter in the ocean? Org. Geochem., 27(5-6): 195–212. Doi: 10.1016/S0146-6380(97)00066-1 |
| Hedges J I, 1992. Global biogeochemical cycles:progress and problems. Mar. Chem., 39(1-3): 67–93. Doi: 10.1016/0304-4203(92)90096-S |
| Hertkorn N, Benner R, Frommberger M, Schmitt-Kopplin P, Witt M, Kaiser K, Kettrup A, Hedges J I, 2006. Characterization of a major refractory component of marine dissolved organic matter. Geochim. Cosmochim. Acta, 70(12): 2 990–3 010. Doi: 10.1016/j.gca.2006.03.021 |
| Jacobs G A, Hur H B, Riedlinger S K, 2000. Yellow and East China Seas response to winds and currents. J. Geophys. Res., 105(C9): 21 947–21 968. Doi: 10.1029/2000JC900093 |
| Kisand V, Gebhardt S, Rullkötter J, Simon M, 2013. Significant bacterial transformation of riverine humic matter detected by pyrolysis GC-MS in serial chemostat experiments. Mar. Chem., 149: 23–31. Doi: 10.1016/j.marchem.2012.12.003 |
| Koch B P, Witt M, Engbrodt R, Dittmar T, Kattner G, 2005. Molecular formulae of marine and terrigenous dissolved organic matter detected by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Geochim. Cosmochim. Acta, 69(13): 3 299–3 308. Doi: 10.1016/j.gca.2005.02.027 |
| Koprivnjak J F, Pfromm P H, Ingall E, Vetter T A, SchmittKopplin P, Hertkorn N, Frommberger M, Knicker H, Perdue E M, 2009. Chemical and spectroscopic characterization of marine dissolved organic matter isolated using coupled reverse osmosis-electrodialysis. Geochim. Cosmochim. Acta, 73(14): 4 215–4 231. Doi: 10.1016/j.gca.2009.04.010 |
| Lam B, Baer A, Alaee M, Lefebvre B, Moser A, Williams A, Simpson A J, 2007. Major structural components in freshwater dissolved organic matter. Environ. Sci. Technol., 41(24): 8 240–8 247. Doi: 10.1021/es0713072 |
| Leenheer J A, 1981. Comprehensive approach to preparative isolation and fractionation of dissolved organic carbon from natural waters and wastewaters. Environ. Sci.Technol., 15(5): 578–587. Doi: 10.1021/es00087a010 |
| Li H M, Shi X Y, Shang R N, Han X R. 2013. Distribution of dissolved organic carbon and its influence factors in the Bohai Sea and Yellow Sea in autumn. Mar. Environ. Sci., 32(2): 161-164, 181. (in Chinese with English abstract) http://en.cnki.com.cn/Article_en/CJFDTotal-HYHJ201302001.htm |
| Mao J D, Cory R M, McKnight D M, Schmidt-Rohr K, 2007. Characterization of a nitrogen-rich fulvic acid and its precursor algae from solid state NMR. Org. Geochem., 38(8): 1 277–1 292. Doi: 10.1016/j.orggeochem.2007.04.005 |
| Mao J D, Fang X W, Lan Y Q, Schimmelmann A, Mastalerz M, Xu L, Schmidt-Rohr K, 2010. Chemical and nanometerscale structure of kerogen and its change during thermal maturation investigated by advanced solid-state 13C NMR spectroscopy. Geochim. Cosmochim. Acta, 74(7): 2 110–2 127. Doi: 10.1016/j.gca.2009.12.029 |
| Mao J D, Kong X Q, Schmidt-Rohr K, Pignatello J J, Perdue E M, 2012. Advanced solid-state NMR characterization of marine dissolved organic matter isolated using the coupled reverse osmosis/electrodialysis method. Environ. Sci.Technol., 46(11): 5 806–5 814. Doi: 10.1021/es300521e |
| Meyers P A, 1994. Preservation of elemental and isotopic source identification of sedimentary organic matter. Chem. Geol., 114(3-4): 289–302. Doi: 10.1016/0009-2541(94)90059-0 |
| Mopper K, Stubbins A, Ritchie J D, Bialk H M, Hatcher P G, 2007. Advanced instrumental approaches for characterization of marine dissolved organic matter:extraction techniques, mass spectrometry, and nuclear magnetic resonance spectroscopy. Chem. Rev., 107(2): 419–442. Doi: 10.1021/cr050359b |
| Nebbioso A, Piccolo A, 2013. Molecular characterization of dissolved organic matter (DOM):a critical review. Anal.Bioanal. Chem., 405(1): 109–124. Doi: 10.1007/s00216-012-6363-2 |
| Perdue E M, Benner R. 2009. Marine organic matter. In: Senesi N, Xing B S, Huang P M eds. Biophysico-Chemical Processes Involving Natural Nonliving Organic Matter in Environmental Systems. John Wiley & Sons, Hoboken, New Jersey. p. 407-449. |
| Reemtsma T, These A, Springer A, Linscheid M, 2006. Fulvic acids as transition state of organic matter:indications from high resolution mass spectrometry. Environ. Sci.Technol., 40(19): 5 839–5 845. Doi: 10.1021/es060318c |
| Simjouw J P, Minor E C, Mopper K, 2005. Isolation and characterization of estuarine dissolved organic matter:comparison of ultrafiltration and C18 solid-phase extraction techniques. Mar. Chem., 96(3-4): 219–235. Doi: 10.1016/j.marchem.2005.01.003 |
| Stevenson F J, 1994. Humus Chemistry:Genesis, Composition, Reactions. John Wiley & Sons, New York. |
| Thurman E M, Malcolm R L, 1981. Preparative isolation of aquatic humic substances. Environ. Sci. Technol., 15(4): 463–466. Doi: 10.1021/es00086a012 |
| Tremblay L, Alaoui G, Léger M N, 2011. Characterization of aquatic particles by direct FTIR analysis of filters and quantification of elemental and molecular compositions. Environ. Sci. Technol., 45(22): 9 671–9 679. Doi: 10.1021/es202607n |
| Woods G C, Simpson M J, Kelleher B P, McCaul M, Kingery W L, Simpson A J, 2010. Online high-performance size exclusion chromatography-nuclear magnetic resonance for the characterization of dissolved organic matter. Environ. Sci. Technol., 44(2): 624–630. Doi: 10.1021/es903042s |
| Xie L P, Wang Z L, Wang B D, Sun X, Sun P X. 2010. Distribution and controlled factor of dissolved organic carbon in Southern Yellow Sea in spring. Mar. Environ. Sci., 29(5): 636-640. (in Chinese with English abstract) http://en.cnki.com.cn/Article_en/CJFDTOTAL-HYHJ201005005.htm |
| Yuan H M, Song J M, Li X G, Li N, Duan L Q, Qu B X, Lu X, Chen X. 2015. Distribution and impact factors of dissolved organic carbon in the Southern Yellow Sea and the Changjiang Estuary in summer. J. Guangxi Acad. Sci., 31(3): 155-160. (in Chinese with English abstract) http://en.cnki.com.cn/Article_en/CJFDTOTAL-GXKX201503001.htm |
| Zhang Y L, Du J Z, Ding X P, Zhang F F, 2016. Comparison study of sedimentary humic substances isolated from contrasting coastal marine environments by chemical and spectroscopic analysis. Environ. Earth Sci., 75(5): 378. Doi: 10.1007/s12665-016-5263-8 |
| Zhang Y L, Du J Z, Peng B, Zhang F F, Zhao X, Zhang J, 2013a. Chemical and spectroscopic characterization of dissolved humic substances in a mangrove-fringed estuary in the eastern coast of Hainan Island, China. Chin. J.Oceanol. Limnol., 31(2): 454–463. Doi: 10.1007/s00343-013-2138-7 |
| Zhang Y L, Du J Z, Zhang F F, Yu Y H, Zhang J, 2011. Chemical characterization of humic substances isolated from mangrove swamp sediments:the Qinglan area of Hainan Island, China. Estuar. Coast. Shelf Sci., 93(3): 220–227. Doi: 10.1016/j.ecss.2010.12.025 |
| Zhang Y L, Du J Z, Zhao X, Wu W S, Peng B, Zhang J, 2014. A multi-proxy study of sedimentary humic substances in the salt marsh of the Changjiang Estuary, China. Estuar.Coast. Shelf Sci., 151: 295–301. Doi: 10.1016/j.ecss.2014.10.007 |
| Zhang Y L, Green N W, Perdue E M, 2013b. Acid-base properties of dissolved organic matter from pristine and oil-impacted marshes of Barataria Bay, Louisiana. Mar.Chem., 155: 42–49. Doi: 10.1016/j.marchem.2013.05.010 |