 2018, Vol. 36
2018, Vol. 36Institute of Oceanology, Chinese Academy of Sciences
Article Information
- LIANG Pengjuan(梁朋娟), LI Shangyong(李尚勇), WANG Kun(王昆), WANG Fang(王芳), XING Mengxin(邢孟欣), HAO Jianhua(郝建华), SUN Mi(孙谧)
- A thermostable serralysin inhibitor from marine bacterium Flavobacterium sp. YS-80-122
- Chinese Journal of Oceanology and Limnology, 36(2): 483-489
- http://dx.doi.org/10.1007/s00343-018-6266-y
Article History
- Received Oct. 13, 2016
- accepted in principle Dec. 26, 2016
- accepted for publication Feb. 22, 2018




2 Laboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266071, China;
3 Shanghai Ocean University, Shanghai 201306, China
Serralysin-type proteases belong to the M10B subfamily of zinc-metallo endopeptidases (http://merops.sanger.ac.uk/). These proteases are secreted by a large number of bacteria, including human pathogens Pseudomonas aeruginosa, Serratia marcescens and Erwinia chrysanthemi (Louis et al., 1998; Hege and Baumann, 2001; Kida et al., 2008). Serralysins are important virulence factors in the development of diseases, such as pneumonia and keratitis (Kida et al., 2007, 2008). Therefore, serralysin inhibitors may be developed into potent drugs for the treatment of these diseases, and should help to avoid the development of antibiotic resistance amongst these pathogenic bacteria (Dhanaraj et al., 1996; Feltzer et al., 2000).
In general, serralysin-secreting bacteria contain a gene in the serralysin operon that codes for a 10-kDa periplasmic protease inhibitor (Létoffé et al., 1989; Kim et al., 1995; Liao and McCallus, 1998). These serralysin inhibitors likely protect the bacterium from adventitious proteolysis during serralysin secretion. Although a large number of serralysin inhibitor genes have been sequenced during whole bacterial genome analysis, there are only a few reports describing the characterization of serralysin inhibitors and their corresponding serralysins. Currently, only three serralysin inhibitors have been characterized: APRin from P. aeruginosa, Inh from E. chrysanthemi and SmaPI from S. marcescens (Létoffé et al., 1989; Baumann et al., 1995; Kim et al., 1995; Bae et al., 1998; Feltzer et al., 2000; Arumugam et al., 2008). Therefore, the discovery and characterization of new serralysin inhibitors is very important.
In our previous study, a typical serralysin metalloprotease (MP) (GenBank accession No. ACY25898) was purified from the marine bacterium Flavobacterium sp. YS-80-122 (Wang et al., 2010; Li et al., 2016). Preliminary attempts at cloning the MP gene indicated that an inhibitor gene, lupI, was located downstream of the MP gene. In this study, we report the cloning, expression and characterization of the inhibitor LupI from Flavobacterium sp. YS-80-122. Our study on this thermostable protein provides a new candidate for the treatment of serralysin-associated infections.
2 MATERIAL AND METHOD 2.1 Bacterial strains and plasmidsThe marine bacterium Flavobacterium sp. YS-80-122 was isolated from sediment of the Yellow Sea and was cultured at 18℃ in GB medium (yeast extract, 2.4%; peptone, 1.2%; NaCl, 2.5%; glycerol 0.4%) (Wang et al., 2010). Escherichia coli strains DH5α and BL21 (DE3) (Novagen, Madison, WI, USA) were cultured at 37℃ in Luria-Bertani (LB) broth supplemented with ampicillin (100 μg/mL). The pET-22b(+) vector (Novagen) was used for gene cloning and expression.
2.2 Cloning and sequence analysis of lupIInitial cloning of the gene coding for MP revealed that an inhibitor gene, lupI, was located downstream of the MP gene. The lupI nucleotide sequence was deposited in the GenBank under the accession number AEO90403.1. A signal peptide was predicted by SignalP 4.1 and comparison analysis was performed using the NCBI conserved domain database. The theoretical molecular weight (Mw) of LupI was calculated using the Compute Mw Tool available at http://us.expasy.org/tools/. A phylogenetic tree was constructed by the neighbor-joining method using MEGA 6.0 software. The reliability of the phylogenetic reconstructions was tested by boot-strapping (1 000 replicates).
2.3 Expression and purification of recombinant LupIFor expression of LupI, a DNA fragment containing lupI without the proposed DNA fragment encoding the signal peptide and stop codon was amplified using primers lupIF (CATGCTTGGCCAGTTCGCTGATGTTATTAAG) and lupIR (CATGTCAAGCTTTTAATGCACACTTTGTAAG) to introduce MscⅠ and HindⅢ sites (underlined), respectively, into lupI. The purified product was ligated into the corresponding sites of the pET22b vector, generating recombinant plasmid pET22b-lupI, which was transformed into chemically competent E. coli BL21 (DE3) cells. The resulting transformants were selected on LB medium supplemented with ampicillin (100 μg/mL). A single transformant was cultured in LB broth supplemented with 100 μg/mL ampicillin at 37℃ to an optical density (600 nm) of 0.6 and the expression of the target gene was then induced by the addition of isopropyl-β-thiogalactoside to a final concentration of 0.1 mmol/L. The culture was incubated at 37℃ and 200 r/min for an additional 6 h. Cells were pelleted by centrifugation at 8 000×g for 20 min at 4℃ and then resuspended in Tris-HCl buffer (50 mmol/L, pH 8.0), and ultrasonically disrupted. Following centrifugation at 10 000×g for 20 min at 4℃, the supernatant was loaded onto a His-Trap HP column (GE Healthcare) equilibrated with buffer I (50 mmol/L Tris-HCl, 100 mmol/L NaCl, pH 8.0). The column was washed using buffer I containing 25 mmol/L imidazole and then eluted with buffer I containing 150 mmol/L imidazole. The protein concentration was measured by the Bradford method, using bovine serum albumin (BSA) as the standard. The molecular weight of the purified LupI was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDSPAGE) on a 12% (w/v) resolving gel.
2.4 Inhibitory activity of LupI against MPThe MP metalloprotease was purified by fast protein liquid chromatography (FPLC) using a Superdex 200 column HR10/30 (Amersham Pharmacia Biotech, Uppsala, Sweden). Protease activity was assayed by measuring the amount of tyrosine liberated from casein. Casein was dissolved in 25 mmol/L borate buffer (pH 10.0) to a concentration of 1% (w/v). The purified MP and casein solutions were pre-incubated at 25℃ for 10 min, and then equal volumes (1 mL) of each were mixed and incubated at 25℃ for 10 min. The amount of released tyrosine was measured as described previously (Hao and Sun, 2015; Li et al., 2016). One unit was defined as the amount of protease releasing 1 μg of tyrosine per min. Inhibition assays were undertaken by mixing purified LupI (0.2 μmol/L) with purified MP (0.4 μmol/L), followed by incubation at 25℃ for 10 min. The activity of the MP was then determined as described above.
2.5 Enzyme kinetics assay to measure LupI inhibition of MPEnzymatic assays were carried out in a buffer containing 50 mmol/L Tris/HCl (pH 8.0), 100 mmol/L NaCl, 10 mmol/L CaCl2, 10 μmol/L ZnCl2 and 0.001% BSA at 25℃. MP was used at a concentration of 0.5 μmol/L and various concentrations of LupI were investigated (0–0.4 μmol/L). N-α-benzoyl-D, L-arginine-4-nitroanilide was used as a substrate to determine the kinetic parameters of the interaction between MP and LupI, as described previously (Bae et al., 1998). The absorbance of the solution was monitored at 405 nm and each experiment was performed in triplicate. The inhibitory constant, Ki, was determined from the Lineweaver-Burk and Dixon equations (Ji et al., 2013). Briefly, the Michaelis constant (Km) of the MP was determined using Lineweaver-Burk plots and the Ki value was obtained from Dixon plots. LupI induced a competitive inhibition. To describe the mechanism of competitive inhibition, the Lineweaver-Burk equation in double reciprocal form was written as:
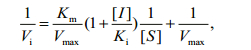 (1)
(1)secondary plots were constructed from the equation:
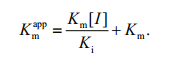 (2)
(2)The Ki, Km and Vmax values were derived from the above equations. The secondary replot of the apparent Km vs. [I] was linearly fitted, assuming a single inhibition site or a single class of inhibition sites.
2.6 Heat tolerance assaysTo determine the heat tolerance of LupI, residual inhibitory activity was determined under standard assay conditions (described in Section 2.5.) after incubating LupI at 100℃ for various periods (1, 5, 10, 15, 30, 60 min). Samples were heat-treated at 100℃ and then immediately incubated at 0℃ for 60 min. To test the effect of temperature on the recovery of heat-treated LupI, the inhibitor was incubated at 100℃ for 10 min and then immediately incubated at temperatures ranging from 0–50℃ for 60 min. The recovered inhibitory activity was then examined under standard conditions. To test the impact of the incubation time on the recovery of heattreated LupI, the inhibitor was incubated at 100℃ for 10 min and then held at 0℃ for various time periods (ranging from 0–60 min). The recovered activity of LupI was tested as described in Section 2.5.
3 RESULT 3.1 Sequence analysis of lupIGenetic analysis of the lupI serralysin inhibitor gene region indicated that the intergenic distance between the stop codon (TAA) of the metalloprotease MP gene and the start codon (ATG) of lupI was only 58 bp. The lupI gene is 363 bp in length and the deduced protein is composed of 121 amino acids (Supplementary Fig. S1). Signal peptide prediction by SignalP 4.1 and comparison analysis using the NCBI conserved domain database indicated that LupI was a single-domain protein containing a putative signal peptide (Met1–Ala17). The theoretical Mw and isoelectric point of the mature enzyme were 11 295 Da and 6.68, respectively.

|
| Figure S1 The coding DNA sequence of lupI |
LupI showed the highest amino acid sequence identity (36.9%) to Inh (GenBank accession no. CAA37341) from E. chrysanthemi, followed by SmaPI (GenBank accession No. L09107) from S. marcescens (31.6% identity) and APRin (GenBank accession No. CRP83773) from P. aeruginosa (28.7% identity). A phylogenetic tree (Fig. 1) was constructed for LupI with other known serralysin and matrix metalloprotease inhibitors. LupI formed a branched cluster with other serralysin inhibitors, including Inh, SmaPI and APRin, in the phylogenetic tree.

|
| Figure 1 Phylogenetic relationship between LupI and other metalloprotease inhibitors Molecular Evolutionary Genetics Analysis (MEGA) version 6.0 was used to construct the phylogenetic tree by the neighbor-joining method. The reliability of the phylogenetic reconstructions was tested by boot-strapping (1 000 replicates). Numbers associated with each of the branches indicate substitution frequencies per amino acid residue. |
Three-dimensional structure analyses of the serralysin-Inh and APR-APRin complexes demonstrated that the N-terminal residues of these inhibitors occupy the substrate-binding cleft of the proteases. The backbone amide groups of the N-terminal residues of the inhibitors chelate the catalytic zinc atom of the proteases (Baumann et al., 1995; Feltzer et al., 2000, 2003; Hege et al., 2001). The ɑ-helix and β-barrel structure of serralysin inhibitors provide an effective stereo-hindrance effect for the N-terminal residues (Bardoel et al., 2012). As shown in the multiple sequence alignment (Fig. 2), residues at the N-terminus (Ser1, Ser2, Leu3, and Leu5), ɑ-helix (Ala9 and Val11) and loop between β-strands 4 and 5 (Pro57, Thr58, Pro59, Apr60) of LupI are highly conserved with other serralysin inhibitors.

|
| Figure 2 Amino acid sequence alignment of LupI with other serralysin inhibitors The alignment was performed using Clustal omega. The conserved N-terminus, ɑ-helix and loop regions between LupI and other serralysin inhibitors are underlined. |
The lupI gene without the proposed DNA region encoding the signal peptide was expressed in E. coli (strain BL21 (DE3)/pET22b-lupI). Recombinant LupI was overexpressed as a soluble product and was purified to homogeneity using His-Trap HP column chromatography. The molecular weight of recombinant LupI was determined to be approximately 11 kDa, which is in agreement with the theoretical molecular weight (Fig. 3).

|
| Figure 3 SDS-PAGE analysis of recombinant LupI Lane M: protein molecular weight markers; lane 1: purified LupI. |
The enzyme kinetics of MP in the presence of LupI was performed using double-reciprocal LineweaverBurk plots. The results (Fig. 4) revealed that while the Vmax values remained constant, the Km values increased with increasing inhibitor concentration, indicating that LupI induced competitive inhibition (Ji et al., 2013). The Km and Vmax values of the MP in the absence of LupI were 5.12 mmol/L and 0.5 fluorescence units, respectively. The Ki of LupI towards the MP was 0.64 μmol/L, according to the Lineweaver-Burk plots.

|
| Figure 4 Lineweaver-Burk plots showing the inhibitory activity of LupI using different concentrations of NαbenzoylD, Larginine4nitroanilide as a substrate The concentrations of purified LupI were 0.4 μmol/L (diamond); 0.2 μmol/L (triangle); 0 μmol/L (dot). |
The thermostability assay showed that the inhibitory activity of LupI recovered 35.6%–90.7% of that of non-heat-treated samples when incubated at 0℃ for 60 min after treatment at 100℃ for various periods (1, 5, 10, 15, 30, 60 min) (Fig. 5). To determine the effect of incubation temperature on the activity of heat-inactivated LupI, samples were incubated at various temperatures (0–50℃) for 60 min immediately after treatment at 100℃ for 10 min. The inhibitory activity of LupI reached 63.5%–70.4% of that of untreated samples when incubated at 0–30℃, but was much lower at temperatures > 30℃ (Fig. 6a). The effect of recovery time post-heat-shock was also investigated and the results showed that the maximum recovered activity (70.4%) was achieved when samples were incubated at 0℃ for 60 min (Fig. 6b).

|
| Figure 5 Analysis of the thermostability of LupI Purified LupI was treated at 100℃ for various periods (1, 5, 10, 15, 30, 60 min) and then immediately incubated at 0℃ for 60 min. The residual inhibitory activity was assayed. Statistical analysis was used to generate the data. The mean values of three replicates with standard deviations are shown. |

|
| Figure 6 Effect of incubation temperature (a) and time (b) on the recovered activity of heat-inactivated LupI The inhibitory activity of the non-treated enzyme was used as the control (100%). The average values of three replicates along with standard deviations are shown. |
The metalloproteinase MP, belonging to the serralysin family, is used in several industrial processes. In this study, lupI, coding for a serralysin inhibitor, was cloned from the downstream region of the Flavobacterium sp. YS-80-122 MP gene. Although the deduced protein, LupI, showed < 40% identity to other reported serralysin inhibitors, the regions essential for interaction between serralysin and the inhibitor were highly conserved (Fig. 2), including residues located in the N-terminal (Ser1, Ser2, Leu3 and Leu5), ɑ-helix (Ala9 and Val11) and loop (Pro57, Thr58, Pro59 and Apr60) regions (Fig. 2). These results indicate that LupI is a novel member of serralysin inhibitor.
The Ki of LupI towards MP was 0.64 μmol/L. The Ki values of the characterized serralysin inhibitors from S. marcescens and E. chrysanthemi against their respective target proteinases were 0.713 μmol/L (Bae et al., 1998) and 1–10 μmol/L (Létoffé et al., 1989), respectively, indicating weaker inhibitory activity than that of LupI.
LupI was thermostable, and retained a high degree of activity (35.6%–90.7%) after treatment at 100℃ for 1–60 min, with a half-life (t1/2) of ~30 min. As documented previously, the other two reported inhibitors are also thermostable. The t1/2 of Inh was > 30 min at 95℃ (Baumann et al., 1995), whereas SmaPI was stable at 100℃ for up to 30 min (Kim et al., 1995).
Based on the stronger inhibitor activity, higher degree of thermo-stability and reversible nature of the damage caused by heat treatment, LupI may represent a better option for the treatment of serralysinassociated infections when compared with that of previously documented serralysin inhibitors.
5 CONCLUSIONIn this report, we have identified a new serralysin inhibitor, LupI, from the marine strain YS-80, which shows < 40% amino acid sequence identity to reported serralysin inhibitors. This inhibitor exhibited stronger interaction with the target MP protease than the other two reported serralysin inhibitors SmapI and Inh with their respective proteases. Moreover, LupI is thermostable and recovered 35.6%–90.7% of its initial activity after treatment at 100℃ for periods ranging between 1 and 60 min, followed by incubation at 0℃ for 60 min. The results suggest that LupI is a suitable option for the treatment of serralysinassociated infections.
| Arumugam S, Gray R D, Lane A N, 2008. NMR structure note:alkaline proteinase inhibitor APRin from Pseudomonas aeruginosa. J. Biomol. NMR, 40(3): 213–217. Doi: 10.1007/s10858-008-9218-6 |
| Bae K H, Kim I C, Kim K S, Shin Y C, Byun S M, 1998. The Leu-3 residue of Serratia marcescens metalloprotease inhibitor is important in inhibitory activity and binding with Serratia marcescens metalloprotease. Arch. Biochem.Biophys., 352(1): 37–43. Doi: 10.1006/abbi.1997.0561 |
| Bardoel B W, van Kessel K P M, van Strijp J A G, Milder F J, 2012. Inhibition of Pseudomonas aeruginosa virulence:characterization of the AprA-AprI interface and species selectivity. J. Mol. Biol., 415(3): 573–583. Doi: 10.1016/j.jmb.2011.11.039 |
| Baumann U, Bauer M, Létoffé S, Delepelaire P, Wandersman C, 1995. Crystal structure of a complex between Serratia marcescens metallo-protease and an inhibitor from Erwinia chrysanthemi. J. Mol. Biol., 248(3): 653–661. Doi: 10.1006/jmbi.1995.0249 |
| Dhanaraj V, Ye Q Z, Johnson L L, Hupe D J, Ortwine D F, Dunbar J B Jr, Rubin J R, Pavlovsky A, Humblet C, Blundell T L, 1996. Designing inhibitors of the metalloproteinase superfamily:comparative analysis of representative structures. Drug Des. Discov., 13(3-4): 3–14. |
| Feltzer R E, Gray R D, Dean W L, Pierce W M Jr, 2000. Alkaline proteinase inhibitor of Pseudomonas aeruginosa:interaction of native and N-terminally truncated inhibitor proteins with Pseudomonas metalloproteinases. J. Biol. Chem., 275(28): 21 002–21 009. Doi: 10.1074/jbc.M002088200 |
| Feltzer R E, Trent J O, Gray R D, 2003. Alkaline proteinase inhibitor of Pseudomonas aeruginosa:a mutational and molecular dynamics study of the role of n-terminal residues in the inhibition of pseudomonas alkaline proteinase. J. Biol. Chem., 278(28): 25 952–25 957. Doi: 10.1074/jbc.M212691200 |
| Hao J H, Sun M, 2015. Purification and characterization of a cold alkaline protease from a psychrophilic Pseudomonas aeruginosa HY1215. Appl. Biochem. Biotechnol., 175(2): 715–722. Doi: 10.1007/s12010-014-1315-2 |
| Hege T, Baumann U, 2001. Protease C of Erwinia chrysanthemi:the crystal structure and role of amino acids Y228 and E189. J. Mol. Biol., 314(2): 187–193. Doi: 10.1006/jmbi.2001.5124 |
| Hege T, Feltzer R E, Gray R D, Baumann U, 2001. Crystal structure of a complex between Pseudomonas aeruginosa alkaline protease and its cognate inhibitor:inhibition by a Zinc-NH2 coordinative bond. J. Biol. Chem., 276(37): 35 087–35 092. Doi: 10.1074/jbc.M104020200 |
| Ji X F, Zheng Y, Wang W, Sheng J, Hao J H, Sun M, 2013. Virtual screening of novel reversible inhibitors for marine alkaline protease MP. J. Mol. Graph. Model., 46: 125–131. Doi: 10.1016/j.jmgm.2013.10.004 |
| Kida Y, Higashimoto Y, Inoue H, Shimizu T, Kuwano K, 2008. A novel secreted protease from Pseudomonas aeruginosa activates NF-κB through protease-activated receptors. Cell. Microbiol., 10(7): 1 491–1 504. Doi: 10.1111/j.1462-5822.2008.01142.x |
| Kida Y, Inoue H, Shimizu T, Kuwano K, 2007. Serratia marcescens serralysin induces inflammatory responses through protease-activated receptor 2. Infect. Immun., 75(1): 164–174. Doi: 10.1128/IAI.01239-06 |
| Kim K S, Kim T U, Kim I J, Byun S M, Shin Y C, 1995. Characterization of a metalloprotease inhibitor protein(SmaPI) of Serratia marcescens. Appl. Environ.Microbiol., 61(8): 3 035–3 041. |
| Létoffé S, Delepelaire P, Wandersman C, 1989. Characterization of a protein inhibitor of extracellular proteases produced by Erwinia chrysanthemi. Mol. Microbiol., 3(1): 79–86. Doi: 10.1111/mmi.1989.3.issue-1 |
| Li S Y, Wang L N, Yang J, Bao J, Liu J Z, Lin S X, Hao J H, Sun M, 2016. Affinity purification of metalloprotease from marine bacterium using immobilized metal affinity chromatography. J. Sep. Sci., 39(11): 2 050–2 056. Doi: 10.1002/jssc.201600104 |
| Liao C H, McCallus D E, 1998. Biochemical and genetic characterization of an extracellular protease from Pseudomonas fluorescens CY091. Appl. Environ.Microbiol., 64(3): 914–921. |
| Louis D, Sorlier P, Wallach J, 1998. Quantitation and enzymatic activity of the alkaline protease from Pseudomonas aeruginosa in culture supernatants from clinical strains. Clin. Chem. Lab. Med., 36(5): 295–298. |
| Wang F, Hao J H, Yang C Y, Sun M, 2010. Cloning, expression, and identification of a novel extracellular cold-adapted alkaline protease gene of the marine bacterium strain YS-80-122. Appl. Biochem. Biotechnol., 162(5): 1 497–1 505. Doi: 10.1007/s12010-010-8927-y |