 2018, Vol. 36
2018, Vol. 36Institute of Oceanology, Chinese Academy of Sciences
Article Information
- WANG Bin(王斌), LIU Hongmei(刘红梅), TANG Haitian(唐海田), HU Xiaoke(胡晓珂)
- Microbial ecological associations in the surface sediments of Bohai Strait
- Chinese Journal of Oceanology and Limnology, 36(3): 795-804
- http://dx.doi.org/10.1007/s00343-018-6289-4
Article History
- Received Nov. 7, 2016
- accepted in principle Feb. 7, 2017
- accepted for publication Mar. 3, 2017




2 University of Chinese Academy of Sciences, Beijing 100049, China;
3 School of Ocean, Yantai University, Yantai 264003, China;
4 Yantai Marine Environment Monitoring Central Station of Oceanic Administration, Yantai 264006, China
Microbial communities are recognized as efficient players in the biogeochemical cycles of carbon, nitrogen, phosphorus, and sulfur (Fuhrman, 2009; Chaffron et al., 2010; Louvado et al., 2015). Investigations of microbial communities in sediments are therefore critical for understanding the marine ecosystem. A fundamental question is the bacterial community assembly, which is simultaneously regulated by stochastic processes (e.g., birth, death and dispersion) and the environmental conditions (Lennon et al., 2012; Coutinho et al., 2015). The environmental variables in a marine system are defined by the hydrological environment (Bi et al., 2011; Dixon et al., 2014).
As straits separate and connect two large bodies of water, they are influenced by strong tidal currents, transportation, and the exchange of various materials (e.g., nutritional chemicals and pollutants) (Li et al., 2015). Deep straits are characterized by stratification, oceanic currents and salinity/density differences on either side, while in shallow straits there are always large tidal currents forced by winds and oceanic inputs (Li et al., 2015). Intense bioturbation, high sedimentation rates, and low deposition of organicrich sediments can also be found in shallow straits (Behbahani et al., 2015). Coastal straits are also strongly influenced by human activities, which exacerbate the environmental complexity. Thus, coastal straits are representatives of complex hydrological environments. Investigations of the bacterial communities in coastal straits will therefore help to understand the microbial community overall in marine ecosystems.
Owing to the advancement of high-throughput DNA sequencing technologies, studies of complex marine microbial communities based on 16S rRNA genes have made great progress in recent years. In Fram Strait, located between Greenland and Svalbard at a depth greater than 3 500 m, marine bacteria in sediments exhibited distinct biogeographic patterns and depth was one of the important variables influencing bacterial diversity (Jacob et al., 2013). Moreover, interactive effects on the degradation activity of marine bacteria were induced by multiple environmental changes in the sediments in a deep strait (Piontek et al., 2015). However, few studies on the bacterial community in shallow straits have been reported.
The Bohai Strait (average depth around 20 m), which separates the Bohai Sea from the Yellow Sea, is adjacent to well-developed cities, Dalian and Yantai in the north and south, respectively, and classified as a shallow strait (Li et al., 2015). The Yellow Sea Warm Current (YSWC), which is a branch of the Tsushima Warm Current, flows into the Bohai Sea mainly in winter through the Bohai Strait. In addition, the Yellow Sea Cold Water Mass (YSCWM) that has cold and high salinity bottom waters partly presents in the northeastern Bohai Strait (Li et al., 2006). Therefore, the Bohai Strait is an ideal model to study coastal and shallow straits. Over the past few years, studies have investigated water circulation (Naimie et al., 2001; Li et al., 2015), suspended sediment transport (Bi et al., 2011), and zoobenthos communities (Mu et al., 2002). However, there are few studies on sediment microbial communities in this region. Thus, evaluating the roles of the sediment bacterial communities in the Bohai Strait is very important for understanding the microbial ecology in coastal and shallow straits overall.
So far, most studies on marine microbial communities have focused on their geographical diversity with emphasis on describing taxonomic composition, alpha/beta diversity, and the influences of environmental variables (Signori et al., 2014; Zheng et al., 2014). In addition to taxonomic composition, investigations on microbial associations should be included to understand the full assembly of bacterial communities (Faust and Raes, 2012; Coutinho et al., 2015). Those co-occurring bacteria may apply different strategies to avoid competition for resources in their environments (Macalady et al., 2008). The co-occurrence patterns among microorganisms can reveal the ecological associations and the key populations within the communities (Chaffron et al., 2010; Faust and Raes, 2012). Comprehensive analysis of the relationship between microbial association networks and related ecological functions will help to elucidate the roles of sediment microbial communities in straits.
In this study, six stations in the Bohai Strait were selected to explore microbial communities in the surface sediments in the summer of 2014. We investigated the taxonomic composition of microbial communities, their ecological associations, the key populations, and the environmental influences.
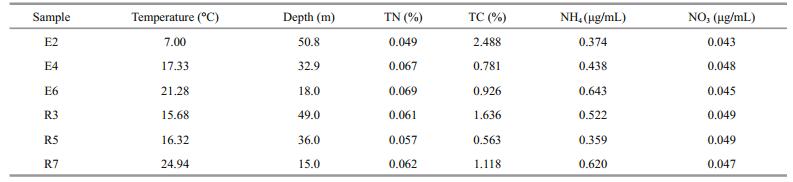
2 MATERIAL AND METHOD 2.1 Survey stations, sediment sampling, and physicochemical analysisSix stations were sampled to cover the Bohai Strait (Fig. 1). The R and E sections represented two sides adjacent to the Bohai and Yellow seas, respectively. Between 27 August and 6 September 2014, composite surface sediment samples (0-5 cm) were collected in triplicate with a box grab at each station. The samples (500 g each) were pooled in sterile polythene bags and stored at -80℃ in the dark until processing. The physical parameters, including temperature, were determined in bottom water on-site using a Sea-Bird 911 plus CTD probe (Sea-Bird Electronics, Bellevue, WA, USA). Total carbon (TC), total nitrogen (TN), and mineral nitrogen (ammonium [NH4+] and nitrate [NO3-]) in the samples were determined after freezedrying, as described by Guan et al. (2014).

|
| Figure 1 The six survey station locations (triangles) across the Bohai Strait (plotted with Surfer 11.0TM) |
To extract genomic DNA, the MoBio PowerSoilTM kit (MoBio, Carlsbad, CA, USA) was used according to the manufacturer's instructions by adding 0.5 g sediment to the kit. For each station, three replicates were extracted and then mixed. Six genomic DNA samples were obtained in total and then sent to Chengdu Institute of Biology, Chinese Academy of Sciences for sequencing analysis. The V4-V5 region of the bacterial 16S rRNA gene using the universal primers 515F (5'-GTGCCAGCMGCCGCGG-3') and 907R (5'-CCGTCAATTCMTTTRAGTTT-3') were sequenced on the Illumina-Miseq platform (Illumina Inc., San Diego, CA, USA). PEAR software (Zhang et al., 2014) was used to merge the paired reads with default settings and QIIME software with default parameters (Caporaso et al., 2010) was applied to process the merged sequences. Reads were filtered according to the quality scores and then split into samples according to the barcode information (split_ libraries.py). Chimeric sequences were identified and removed using UCHIME (identify_chimeric_seqs.py and filter_fasta.py) (Edgar et al., 2011). All samples without chimeric sequences were normalized to the same sequencing depth level at 18 000 sequences. Operational taxonomic units (OTUs) were clustered with a 90% similarity cutoff (Barberán et al., 2012) by using the strategy "pick_closed_reference" and UCLUST (Edgar, 2010). The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed using Greengene (version 13.8, http://www.greengene.com/). Those OTUs unknown at the kingdom level were removed.
2.3 Statistical analysisThe coverage and alpha diversity of the samples were calculated in QIIME on the filtered OTUs. Alpha diversity was estimated using richness (Chao1) and diversity (Shannon) indices to represent community richness and diversity. The "vegan" packages (Dixon, 2003) in R were applied for representational difference analysis (RDA). To identify the metabolic or other functional capabilities of bacteria, a phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) (Langille et al., 2013) was used. This predicted the functional composition of a metagenome by using marker gene data and a database of reference genomes. Following an online tutorial (http://picrust.github.io/picrust/index.html), metagenome predictions against KEGG Ortholog (Ogata et al., 1999; Okuda et al., 2008; Langille et al., 2013) were carried out based on the 16S rRNA sequence dataset. Then, thousands of predicted functions (KOs) were collapsed into higher categories (KEGG Pathways). The category "Energy Metabolism" was selected for further discussion of the ecological functions of microbial communities. Variance analysis was executed in R, with P < 0.05 considered to be statistically significant.
Network analysis was applied to visualize interactions within the sedimentary microbial community (Faust and Raes, 2012; Lupatini et al., 2014). Taxon assignment on lower taxonomic levels is not always accurate, which can confuse ecological signals and artifacts (Faust and Raes, 2012), so we only considered links and patterns above a certain taxonomic rank (Freilich et al., 2010). In this study, 647 (21.8%) and 1881 (63.5%) OTUs could be classified at the genus and family levels, respectively. Therefore, we considered ecological associations at family levels. In order to focus on the common bacteria in the survey region, those OTUs classified at family level and present in more than three samples were selected for the network analysis. Spearman correlations among the OTUs (R > 0.9, P < 0.05, adjusted with "Benjamini-Hochberg") were calculated in R to investigate the associations. After calculation, the associations were summarized at family level. The number of OTU associations (positive or negative) between two families was defined and termed NC, which is an estimate of the strength of the associations at the family level. Then, the positive and negative networks with weighted connections (NC values) were constructed using Cytoscape 3.2.1 (Smoot et al., 2011).
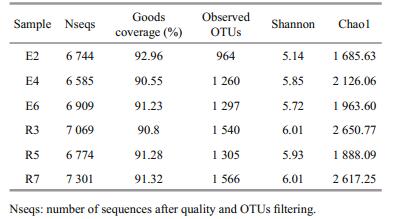
3 RESULT 3.1 Environmental conditions, bacterial communities and prediction metagenomeThis study covered six stations in two regions (R and E) of the Bohai Strait in northern China (Fig. 1). The environmental variables are summarized in Table 1. The depths ranged from 15 m (station R7) to 50.8 m (E2), with temperatures from 7℃ (E2) to 24.94℃ (R7). The TC content in E2, R3, E6 and R7 were higher than in E4 and R5. A total of 41382 16S rRNA gene sequences were obtained across all samples after quality and OTU filtering. Good's coverage in all samples were greater than 90%, indicating that sequencing captured the majority of the bacterial diversity in the samples (Table 1). The average Shannon and Chao1 values in the R section (R3, R5 and R7, Meanshannon=5.98±0.04, MeanChao1=2 385.37±351.90) were higher than in the E section (E2, E4 and E6, Meanshannon=5.57±0.31, Meanchao1=1 925.10±181.85). The highest diversity (Shannon=6.01) and richness (Chao1=2 650.77) values were found in R3 (Table 2).
|
In total, 2962 OTUs affiliated to 140 classes with 204 families were identified. At the class level, the four most abundant classes were Gammaproteobacteria (19.34%±5.02%), Deltaproteobacteria (16.72%± 5.06%), Bacilli (12.07%±2.05%), and Flavobacteriia (11.08%±5.07%). However, the relative abundance of the Flavobacteriia was lower in R3 (3.14%) than in other samples (Fig. 2a). At the family level, Lactobacillaceae (10.55%±1.89%) and Flavobacteriaceae (10.46%±4.94%) were the most abundant families. Psychromonadaceae represented a higher relative abundance in E2 (9.02%) than in other samples (Fig. 2b).

|
| Figure 2 Heatmap showing the relative abundance of (a) abundant classes (>1%) and (b) abundant families (>1%) Both samples and phylotype were clustered. |
RDA was applied to investigate the major environmental influences in the Bohai Strait. In total, only 66.6% of the variation could be explained by the first two dimensionalities (Fig. 3). For the first dimensionality, temperature (R=-0.667 4, P < 0.05), TC (R=0.641, P < 0.05) and nitrate (R=-0.918 8, P < 0.05) exhibited significant correlations with RDA1. For RDA2, depth (R=0.749 6, P < 0.05) and TC (R=0.621 6, P < 0.05) were significantly correlated. Twenty-seven summary functions (e.g., energy metabolism, xenobiotic biodegradation, and metabolism), including 234 KEGG pathways (e.g., carbon fixation in prokaryotes), were predicted with PICRUSt. According to the variation of the KEGG pathways (Fig. 4a), Paths 2 (carbon fixation in prokaryotes), 3 (methane metabolism), 5 (oxidative phosphorylation) and 9 (sulfur metabolism) were relatively varied among samples. Nitrate (R=-0.948 8, P < 0.05) and TC (R=0.77, P < 0.05) were significantly correlated with RDA2 (Fig. 4b).

|
| Figure 3 The relationship between bacterial communities and environmental variables from RDA T: temperature; TC: total carbon; TN: total nitrogen; NO3: nitrate; NH4: ammonium. |

|
| Figure 4 Summary of predicted KEGG pathways of energy metabolism a. box plots showing variations of the relative abundance of different pathways across six samples; b. the relationship between nine pathways and the environmental variables from RDA: Path 1: carbon fixation in photosynthetic organisms; Path 2: carbon fixation in prokaryotes; Path 3: methane metabolism; Path 4: nitrogen metabolism; Path 5: oxidative phosphorylation; Path 6: photosynthesis; Path 7: photosynthesis - antenna proteins; Path 8: photosynthesis proteins; Path 9: sulfur metabolism. T: temperature; TC: total carbon; TN: total nitrogen; NO3: nitrate; NH4: ammonium. |
The positive (P-network) and negative (N-network) networks were constructed to demonstrate microbial associations at the family level. The P-network consisted of 115 nodes and 962 edges (Fig. 5a) while 108 nodes and 750 edges were found in the N-network (Fig. 6a). The heterogeneity of both networks were severe. To reduce the complexity of the networks, a strong association was defined as a connection with a NC value greater than 5 and then two new networks with strong associations were extracted (Figs. 5b, 6b). The extracted positive network (EP-network) was comprised of 51 nodes and 118 edges (Fig. 5b). In the EP-network, the five most connected families were Flavobacteriaceae (27 connections), Piscirickettsiaceae (23 connections), Pirellulaceae (20 connections), Desulfobulbaceae (17 connections), and OM60 (16 connections). The four strongest associations were found between Piscirickettsiaceae and Desulfobulbaceae (NC=42), Flavobacteriaceae and Desulfobulbaceae (NC=35), OM60 and Flavobacteriaceae (NC=34), and Desulfobacteraceae and Flavobacteriaceae (NC=33). In the extracted negative network (EN-network), Flavobacteriaceae (32 connections), Piscirickettsiaceae (13 connections), Chitinophagaceae (6 connections), Desulfobacteraceae (6 connections), and Desulfobulbaceae (6 connections) were the five most connected families (Fig. 6b).

|
| Figure 5 Network visualization diagrams showing correlations at the family level for the positive associations The NC value was equal to the number of OUT associations between two families. a. the positive network; b. the extracted network with NC edge values >5. |

|
| Figure 6 Network visualization diagrams showing correlations at the family level for negative associations a. the negative network; b the extracted network with NC edge values >5. The NC value was equal to the number of OTU associations between two families. The width and color of edges represents the NC values. |
One fundamental question was how the environmental variables shape the structure of the microbial communities. The average Chao1 and Shannon values were both higher in the R region of the Bohai Strait sampled in this study (Table 2). Additionally, both of the Shannon and Chao1 indices were higher in the Bohai Sea than reported from the northern Yellow Sea (Liu et al., 2015). This suggests that the adjacent seas influence the microbial communities in the Bohai Strait. The niche theory postulates that the abundance and composition of microbial communities are determined entirely by the environmental conditions (Nemergut et al., 2013; Coutinho et al., 2015). In previous studies, different environmental factors affecting microbial communities were observed in different survey regions in the Bohai Sea. The median grain size and dissolved oxygen were the most important in intertidal sediments (Wang et al., 2013), while TN and total organic carbon were the most important in coastal sediments (Guan et al., 2014; Wang et al., 2015). However, temperature was always important (Wang et al., 2013, 2015; Liu et al., 2015). In this study, temperature, TC, depth and NO3- were the major factors responsible for the dissimilarity of the sediment microbial communities in the survey region (Fig. 3). Similarly, comparisons of Bohai Bay (Guan et al., 2014), Laizhou Bay (Wang et al., 2014), and Liaodong Bay (Zheng et al., 2014) indicated that temperature and nitrate were important parameters affecting sediment bacterial abundance and diversity in the Bohai Sea.
Another question was about ecological function and the related microbes. According to the KEGG pathways (Kanehisa et al., 2017), Path 2 (carbon fixation in prokaryotes) was known to exist in autotrophic bacteria and archaea including: (1) microaerophiles and anaerobes such as green sulfur bacteria, (2) strictly anaerobic bacteria and archaea (Proteobacteria, Planctomycetes, Spirochaetes, and Euryarchaeota), (3) some green non-sulfur bacteria of the family Chloroflexaceae, and (4) aerobic Crenarchaeota, Acidianus, Metallosphaera, and Sulfolobales. Path 3 is principally implemented by methanotrophs and methanogens. In this study, the class of Ignavibacteria with the highest relative abundance (0.54%, data not shown) in R3 was linked to Path 2. Path 3-related microbes might consist of Methanobacteria and Methanomicrobia and the family Methanomassiliicoccaceae. Nitrate and TC content can explain the variations in the nine KEGG pathways of energy metabolism (Fig. 4b). Moreover, terrigenous nutrient input (mainly nitrogen from the wide use of nitrogen-based fertilizer) and material exchange between the Bohai and Yellow seas may lead to different nutrient distributions (Zhang et al., 2004), indicating that variations in environmental variables induced by the complex hydrological conditions will exert influences on the microbial communities and their ecological functions in the Bohai Strait.
Microbes with high abundance are known to exert great influence on the structure and ecological function of the overall microbial community (Campbell and Kirchman, 2013, Zhang et al., 2013). Therefore, the shifts in the relative abundance of predominant microbes under different environmental conditions can reflect the adaptations of microbial communities to habitat filtering. The most abundant OTUs (8.33%±1.51%, data not shown) in the family Lactobacillaceae were affiliated with Lactococcus piscium, first isolation from salmonid fish (Sakala et al., 2002). Piscirickettsiaceae are generally considered to be pathogens causing rickettsial disease in fish and have high nutritional needs (Fryer and Hedrick, 2003); these were particularly abundant at the R7 stations (8.51%, Fig. 2b) where a fish farm is located. This implies that fishery breeding affects the microbial communities in surface sediments in the Bohai Strait. Psychromonadaceae only contains the genus Psychromonas and is usually considered a psychrophile with widespread distribution in deep seas, such as the Japan Trench, and Antarctic environments (Maruyama et al., 2000). The relative abundance of this family was higher in the E2 stations (9.02%, Fig. 2b) that have much lower temperatures (Table 1), indicating that the YSCWM influences the bacterial communities in the Bohai Strait. Desulfobacteraceae and Desulfobulbaceae were highly abundant (3.33%±1.21% and 4.85%±0.51%, respectively) across all samples, except at station E2 (Fig. 2b). These families have been previously found in marine sediments and mainly consist of sulfate reducers that completely oxidize organic matter (Elsabé et al., 2012; Na et al., 2015). The lowest relative abundances of the two families in E2 (1.72% and 1.59%, Fig. 2b) suggest that they are intolerant of cold water (Korneeva et al., 2015).
4.2 Key microbial familiesWe studied the interactions among different taxa within sediment microbial communities. Bacteria form complex ecological webs (Faust and Raes, 2012), and the associations are important elements for the structure of microbial communities. The interaction types encompass positive, negative, and neutral impacts. For instance, syntrophy between two species results in benefits to both, a positive impact, while competition between two species results in a negative impact (Hibbing et al., 2010; Molloy, 2014). These interactions can be reflected in their abundance correlations (Faust and Raes, 2012). Numerous studies on microbial biogeography (Campbell and Kirchman, 2013; Jacob et al., 2013; Buttigieg and Ramette, 2015; Coveley et al., 2015; Piontek et al., 2015) and the niche theory (Pontarp et al., 2012) based on habitat filtering have indicated that the members with positive correlations respond similarly to environmental conditions in their habitats. That is, if two species experience a positive correlation in their abundance across multiple environments or samples, this can be interpreted as evidence that they occupy similar ecological niches (Coutinho et al., 2015). Based on these observations, significant correlations between two species were often related to the direct or indirect interactions between them (Barberán et al., 2012; Faust and Raes, 2012; Lupatini et al., 2014; Coutinho et al., 2015). Therefore, connections with high NC values (NC > 5) could explain the strong interactions between two families, for example, Piscirickettsiaceae-Desulfobulbaceae (in the EP-network: NC=42), FlavobacteriaceaeDesulfobulbaceae (NC=35) and OM60- Flavobacteriaceae (NC=34). The key families include Flavobacteriaceae, Pirellulaceae and Piscirickettsiaceae (Figs. 5, 6).
Meanwhile, both the positive and negative networks are scale-free networks preferred by most biological systems (Boccaletti et al., 2006). One distinctive characteristic of the scale-free network is high fault tolerance (Albert et al., 2000). In other words, the forming of an ecological network is crucial for the stability of a microbial ecosystem (Faust and Raes, 2012). On the one hand, the number of connections to one node is the node's degree, and nodes with high degree values are known as hub nodes, which reduce the robustness of the network (Albert et al., 2000; Boccaletti et al., 2006) because the networks are destroyed immediately when the hub nodes are removed. On the other hand, the microbial ecosystem rapidly becomes stable when exogenetic pollutants are introduced into their habitat. For example, our previous study showed that the relative abundances of most microbes under the stress of hydrocarbons with different structures become stable after 30-day incubation (Wang et al., 2016). In this study, the families with high degree values were Flavobacteriaceae, Piscirickettsiaceae and Rhodobacteraceae in both the EP-network and ENnetwork (Figs. 5b, 6b). Flavobacteriaceae represent a major branch of the gram-negative phylum and has undergone rapid expansion incorporating many new species and genera (Bowman, 2006). There are seven phylogenetically related genera in Piscirickettsiaceae, but these genera have very different phenotypic characteristics (Marshall et al., 2014). Rhodobacteraceae exhibits global distribution and about 100 genera are currently recognized (Pujalte et al., 2014). The three key families have high phylogenetic diversity and wide distributions. The high phylogenetic diversity may imply high overlap of the ecological functions within the community. If some microbes in the three families that have certain ecological functions were removed, other microbes could perform similar functions accordingly. Our findings suggest that an important strategy to maintain the stability of the microbial ecosystem is to select families with high phylogenetic diversity as key populations in the microbial association network.
5 CONCLUSIONIn this study, surface sediment bacterial communities at six sites across the Bohai Strait were analyzed in August-September 2014. Investigations into the relationship between microbial communities and environmental variables showed that temperature, total carbon, depth and nitrate were the major influences on the communities. Based on network theory and analysis, the families that are crucial for the stability and functionality of the microbial ecosystem were Flavobacteriaceae, Pirellulaceae and Piscirickettsiaceae.
6 ACKNOWLEDGEMENTThe manuscript was kindly revised by Dr. Shiliang Anthony Liu from Louisiana State University.
Albert R, Jeong H, Barabási A L. 2000. Error and attack tolerance of complex networks. Nature, 406(6794): 378-382. DOI:10.1038/35019019 |
Barberán A, Bates S T, Casamayor E O, Fierer N. 2012. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J., 6(2): 343-351. DOI:10.1038/ismej.2011.119 |
Behbahani R, Hosseinyar G, Lak R. 2015. The controlling parameters on organic matter preservation within the bottom sediments of the northern part of the Persian Gulf. Neues Jahrb. Geol. Pal?ontol. A bh., 276(3): 267-283. DOI:10.1127/njgpa/2015/0485 |
Bi N S, Yang Z S, Wang H J, Fan D J, Sun X X, Lei K. 2011. Seasonal variation of suspended-sediment transport through the southern Bohai Strait. Estuar. Coast. Shelf Sci., 93(3): 239-247. DOI:10.1016/j.ecss.2011.03.007 |
Boccaletti S, Latora V, Moreno Y, Chavez M, Hwang D U. 2006. Complex networks: structure and dynamics. Physics Reports, 424(4-5): 175-308. DOI:10.1016/j.physrep.2005.10.009 |
Bowman J P. 2006. The marine clade of the family flavobacteriaceae: the genera aequorivita, arenibacter, cellulophaga, croceibacter, formosa, gelidibacter, gillisia, maribacter, mesonia, muricauda, polaribacter, psychroflexus, psychroserpens, robiginitalea, salegentibacter, tenacibaculum, ulvibacter, vitellibacter and zobellia. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K H, Stackebrandt E eds. The Prokaryotes: Volume 7: Proteobacteria: Delta, Epsilon Subclass. Springer, New York. p. 677-694.
|
Buttigieg P L, Ramette A. 2015. Biogeographic patterns of bacterial microdiversity in Arctic deep-sea sediments (HAUSGARTEN, Fram Strait). Front. Microbiol., 5: 660. |
Campbell B J, Kirchman D L. 2013. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J., 7(1): 210-220. DOI:10.1038/ismej.2012.93 |
Caporaso J G, Kuczynski J, Stombaugh J, Bittinger K, Bushman F D, Costello E K, Fierer N, Peña A G, Goodrich J K, Gordon J I, Huttley G A, Kelley S T, Knights D, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knights R, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods, 7(5): 335-336. DOI:10.1038/nmeth.f.303 |
Chaffron S, Rehrauer H, Pernthaler J, von Mering C. 2010. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Research, 20(7): 947-959. DOI:10.1101/gr.104521.109 |
Coutinho F H, Meirelles P M, Moreira A P B, Paranhos R P, Dutilh B E, Thompson F L. 2015. Niche distribution and influence of environmental parameters in marine microbial communities: a systematic review. PeerJ, 3: e1008. DOI:10.7717/peerj.1008 |
Coveley S, Elshahed M S, Youssef N H. 2015. Response of the rare biosphere to environmental stressors in a highly diverse ecosystem (Zodletone spring, OK, USA). PeerJ, 3: e1182. DOI:10.7717/peerj.1182 |
Dixon J L, Osburn C L, Paerl H W, Peierls B L. 2014. Seasonal changes in estuarine dissolved organic matter due to variable flushing time and wind-driven mixing events. Estuar. Coast Shelf S ci., 151: 151-210. |
Dixon P. 2003. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science, 14(6): 927-930. DOI:10.1111/j.1654-1103.2003.tb02228.x |
Edgar R C, Haas B J, Clemente J C, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16): 2194-2200. DOI:10.1093/bioinformatics/btr381 |
Edgar R C. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19): 2460-2461. DOI:10.1093/bioinformatics/btq461 |
Elsabé M J, Brüchert V, Fuchs B M. 2012. Vertical shifts in the microbial community structure of organic-rich Namibian shelf sediments. African Journal of Microbiology Research, 6(17): 3887-3897. |
Faust K, Raes J. 2012. Microbial interactions: from networks to models. Nat. Rev. Microbiol., 10(8): 538-550. DOI:10.1038/nrmicro2832 |
Freilich S, Kreimer A, Meilijson I, Gophna U, Sharan R, Ruppin E. 2010. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Research, 38(12): 3857-3868. DOI:10.1093/nar/gkq118 |
Fryer J L, Hedrick R P. 2003. Piscirickettsia salmonis: a Gramnegative intracellular bacterial pathogen of fish. J. Fish Dis., 26(5): 251-262. DOI:10.1046/j.1365-2761.2003.00460.x |
Fuhrman J A. 2009. Microbial community structure and its functional implications. Nature, 459(7244): 193-199. DOI:10.1038/nature08058 |
Guan X Y, Zhu L L, Li Y X, Xie Y X, Zhao M Z, Luo X M. 2014. Composition and variation of sediment bacterial and nirS-harboring bacterial communities at representative sites of the Bohai Gulf coastal zone, China. World J. Microb iol. Biot echnol., 30(4): 1291-1300. DOI:10.1007/s11274-013-1553-4 |
Hibbing M E, Fuqua C, Parsek M R, Peterson S B. 2010. Bacterial competition: surviving and thriving in the microbial jungle. Nature Reviews Microbiology, 8(1): 15-25. DOI:10.1038/nrmicro2259 |
Jacob M, Soltwedel T, Boetius A, Ramette A. 2013. Biogeography of deep-sea benthic bacteria at regional scale (LTER HAUSGARTEN, Fram Strait, Arctic). PLoS One, 8(9): e72779. DOI:10.1371/journal.pone.0072779 |
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research, 45(D1): D353-D361. DOI:10.1093/nar/gkw1092 |
Korneeva V A, Pimenov N V, Krek A V, Tourova T P, Bryukhanov A L. 2015. Sulfate-reducing bacterial communities in the water column of the Gdansk Deep (Baltic Sea). Microbiology, 84(2): 268-277. DOI:10.1134/S002626171502006X |
Langille M G I, Zaneveld J, Caporaso J G, McDonald D, Knights D, Reyes J A, Clemente J C, Burkepile D E, Vega Thurber R L, Knight R, Beiko R G, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotech., 31(9): 814-821. DOI:10.1038/nbt.2676 |
Lennon J T, Aanderud Z T, Lehmkuhl B K, Schoolmaster D R Jr. 2012. Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology, 93(8): 1867-1879. DOI:10.1890/11-1745.1 |
Li G X, Han X B, Yue S H, Wen G Y, Yang R M, Kusky T M. 2006. Monthly variations of water masses in the East China Seas. Cont. Shelf Res., 26(16): 1954-1970. DOI:10.1016/j.csr.2006.06.008 |
Li Y F, Wolanski E, Zhang H. 2015. What processes control the net currents through shallow straits? A review with application to the Bohai Strait, China. Estuar. Coast. Shelf S ci., 158: 1-11. DOI:10.1016/j.ecss.2015.03.013 |
Liu J W, Liu X S, Wang M, Qiao Y L, Zheng Y F, Zhang X H. 2015. Bacterial and archaeal communities in sediments of the North Chinese marginal seas. Microbial. Ecol., 70(1): 105-117. DOI:10.1007/s00248-014-0553-8 |
Louvado A, Gomes N C M, Simões M M Q, Almeida A, Cleary D F R, Cunha A. 2015. Polycyclic aromatic hydrocarbons in deep sea sediments: microbe-pollutant interactions in a remote environment. Sci. Total Env ron., 526: 526-312. |
Lupatini M, Suleiman A K A, Jacques R J S, Antoniolli Z I, de Siqueira Ferreira A, Kuramae E E, Roesch L F W. 2014. Network topology reveals high connectance levels and few key microbial genera within soils. Front. Env ron. Sci., 2: 10. |
Macalady J L, Dattagupta S, Schaperdoth I, Jones D S, Druschel G K, Eastman D. 2008. Niche differentiation among sulfur-oxidizing bacterial populations in cave waters. ISME J., 2(6): 590-601. DOI:10.1038/ismej.2008.25 |
Marshall S H, Gómez F A, Klose K E. 2014. The Genus Piscirickettsia. In: Rosenberg E, DeLong E F, Lory S, Stackebrandt E, Thompson F eds. The Prokaryotes: Gammaproteobacteria. Springer, Berlin Heidelberg. p. 565-573.
|
Maruyama A, Honda D, Yamamoto H, Kitamura K, Higashihara T. 2000. Phylogenetic analysis of psychrophilic bacteria isolated from the Japan Trench, including a description of the deep-sea species Psychrobacter pacificensis sp. nov. Int. J. Syst. Evol. Microbiol., 50(2): 835-846. DOI:10.1099/00207713-50-2-835 |
Molloy S. 2014. Environmental microbiology: disentangling syntrophy. Nature Reviews Microbiology, 12(1): 7. |
Mu F H, Somerfield P J, Warwick R M, Zhang Z N. 2002. Large-scale spatial patterns in the community structure of benthic harpacticoid copepods in the Bohai Sea, China. The Raffles Bulletin of Zoology, 50(1): 17-26. |
Na H, Lever M A, Kjeldsen K U, Schulz F, Jørgensen B B. 2015. Uncultured Desulfobacteraceae and crenarchaeotal group C3 incorporate 13C-acetate in coastal marine sediment. Environ. Microbiol. Rep., 7(4): 614-622. DOI:10.1111/emi4.2015.7.issue-4 |
Naimie C E, Blain C A, Lynch D R. 2001. Seasonal mean circulation in the Yellow Sea—a model-generated climatology. Cont. Shelf Res., 21(6-7): 667-695. DOI:10.1016/S0278-4343(00)00102-3 |
Nemergut D R, Schmidt S K, Fukami T, O'Neill S P, Bilinski T M, Stanish L F, Knelman J E, Darcy J L, Lynch R C, Wickey P, Ferrenberg S. 2013. Patterns and processes of microbial community assembly. Microbiology and Molecular Biology Reviews, 77(3): 342-356. DOI:10.1128/MMBR.00051-12 |
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. 1999. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research, 27(1): 29-34. DOI:10.1093/nar/27.1.29 |
Okuda S, Yamada T, Hamajima M, Itoh M, Katayama T, Bork P, Goto S, Kanehisa M. 2008. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Research, 36(S2): W423-W426. |
Piontek J, Sperling M, Nöthig E M, Engel A. 2015. Multiple environmental changes induce interactive effects on bacterial degradation activity in the Arctic Ocean. Limnology and Oceanography, 60(4): 1392-1410. DOI:10.1002/lno.10112 |
Pontarp M, Canbäck B, Tunlid A, Lundberg P. 2012. Phylogenetic analysis suggests that habitat filtering is structuring marine bacterial communities across the globe. Microbial. Ecol., 64(1): 8-17. |
Pujalte M J, Lucena T, Ruvira M A, Arahal D R, Macián M C. 2014. The family rhodobacteraceae. In: Rosenberg E, DeLong E F, Lory S, Stackebrandt E, Thompson F eds. The Prokaryotes: Alphaproteobacteria and Betaproteobacteria. Springer, Berlin Heidelberg. p. 439-512.
|
Sakala R M, Hayashidani H, Kato Y, Kaneuchi C, Ogawa M. 2002. Isolation and characterization of Lactococcus piscium strains from vacuum-packaged refrigerated beef. Journal of Applied Microbiology, 92(1): 173-179. DOI:10.1046/j.1365-2672.2002.01513.x |
Signori C N, Thomas F, Enrich-Prast A, Pollery R C G, Sievert S M. 2014. Microbial diversity and community structure across environmental gradients in Bransfield Strait, Western Antarctic Peninsula. Front. Microbiol., 5: 647. |
Smoot M E, Ono K, Ruscheinski J, Wang P L, Ideker T. 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics, 27(3): 431-432. |
Wang H, Wang B, Dong W W, Hu X K. 2016. Co-acclimation of bacterial communities under stresses of hydrocarbons with different structures. Sci. Rep., 6: 34588. DOI:10.1038/srep34588 |
Wang L P, Liu L S, Zheng B H, Zhu Y Z, Wang X. 2013. Analysis of the bacterial community in the two typical intertidal sediments of Bohai Bay, China by pyrosequencing. Mar. Pollut. Bull., 72(1): 181-187. DOI:10.1016/j.marpolbul.2013.04.005 |
Wang L P, Zheng B H, Lei K. 2015. Diversity and distribution of bacterial community in the coastal sediments of Bohai Bay, China. Acta Oceanologica Sinica, 34(10): 122-131. DOI:10.1007/s13131-015-0719-3 |
Wang L P, Zheng B H, Nan B X, Hu P L. 2014. Diversity of bacterial community and detection of nirS-and nirK- encoding denitrifying bacteria in sandy intertidal sediments along Laizhou Bay of Bohai Sea, China. Mar. Pollut. Bull., 88(1-2): 215-223. DOI:10.1016/j.marpolbul.2014.09.002 |
Wu H B, Guo Y T, Wang G H, Dai S K, Li X. 2011. Composition of bacterial communities in deep-sea sediments from the South China Sea, the Andaman Sea and the Indian Ocean. African Journal of Microbiology Research, 5(29): 5273-5283. |
Zhang J J, Kobert K, Flouri T, Stamatakis A. 2014. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics, 30(5): 614-620. DOI:10.1093/bioinformatics/btt593 |
Zhang J, Yu Z G, Raabe T, Liu S M, Starke A, Zou L, Gao H W, Brockmann U. 2004. Dynamics of inorganic nutrient species in the Bohai seawaters. J. Mar. Syst., 44(3-4): 189-212. DOI:10.1016/j.jmarsys.2003.09.010 |
Zhang X M, Liu W, Schloter M, Zhang G M, Chen Q S, Huang J H, Li L H, Elser J J, Han X G. 2013. Response of the abundance of key soil microbial nitrogen-cycling genes to multi-factorial global changes. PLoS One, 8(10): e76500. DOI:10.1371/journal.pone.0076500 |
Zheng B H, Wang L P, Liu L S. 2014. Bacterial community structure and its regulating factors in the intertidal sediment along the Liaodong Bay of Bohai Sea, China. Microbiol. Res., 169(7-8): 585-592. DOI:10.1016/j.micres.2013.09.019 |
| Albert R, Jeong H, Barabási A L, 2000. Error and attack tolerance of complex networks. Nature, 406(6794): 378–382. Doi: 10.1038/35019019 |
| Barberán A, Bates S T, Casamayor E O, Fierer N, 2012. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J., 6(2): 343–351. Doi: 10.1038/ismej.2011.119 |
| Behbahani R, Hosseinyar G, Lak R, 2015. The controlling parameters on organic matter preservation within the bottom sediments of the northern part of the Persian Gulf. Neues Jahrb. Geol. Pal?ontol. A bh., 276(3): 267–283. Doi: 10.1127/njgpa/2015/0485 |
| Bi N S, Yang Z S, Wang H J, Fan D J, Sun X X, Lei K, 2011. Seasonal variation of suspended-sediment transport through the southern Bohai Strait. Estuar. Coast. Shelf Sci., 93(3): 239–247. Doi: 10.1016/j.ecss.2011.03.007 |
| Boccaletti S, Latora V, Moreno Y, Chavez M, Hwang D U, 2006. Complex networks: structure and dynamics. Physics Reports, 424(4-5): 175–308. Doi: 10.1016/j.physrep.2005.10.009 |
| Bowman J P. 2006. The marine clade of the family flavobacteriaceae: the genera aequorivita, arenibacter, cellulophaga, croceibacter, formosa, gelidibacter, gillisia, maribacter, mesonia, muricauda, polaribacter, psychroflexus, psychroserpens, robiginitalea, salegentibacter, tenacibaculum, ulvibacter, vitellibacter and zobellia. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K H, Stackebrandt E eds. The Prokaryotes: Volume 7: Proteobacteria: Delta, Epsilon Subclass. Springer, New York. p. 677-694. |
| Buttigieg P L, Ramette A, 2015. Biogeographic patterns of bacterial microdiversity in Arctic deep-sea sediments (HAUSGARTEN, Fram Strait). Front. Microbiol., 5: 660. |
| Campbell B J, Kirchman D L, 2013. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J., 7(1): 210–220. Doi: 10.1038/ismej.2012.93 |
| Caporaso J G, Kuczynski J, Stombaugh J, Bittinger K, Bushman F D, Costello E K, Fierer N, Peña A G, Goodrich J K, Gordon J I, Huttley G A, Kelley S T, Knights D, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knights R, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knight R, 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods, 7(5): 335–336. Doi: 10.1038/nmeth.f.303 |
| Chaffron S, Rehrauer H, Pernthaler J, von Mering C, 2010. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Research, 20(7): 947–959. Doi: 10.1101/gr.104521.109 |
| Coutinho F H, Meirelles P M, Moreira A P B, Paranhos R P, Dutilh B E, Thompson F L, 2015. Niche distribution and influence of environmental parameters in marine microbial communities: a systematic review. PeerJ, 3: e1008. Doi: 10.7717/peerj.1008 |
| Coveley S, Elshahed M S, Youssef N H, 2015. Response of the rare biosphere to environmental stressors in a highly diverse ecosystem (Zodletone spring, OK, USA). PeerJ, 3: e1182. Doi: 10.7717/peerj.1182 |
| Dixon J L, Osburn C L, Paerl H W, Peierls B L, 2014. Seasonal changes in estuarine dissolved organic matter due to variable flushing time and wind-driven mixing events. Estuar. Coast Shelf S ci., 151: 151–210. |
| Dixon P, 2003. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science, 14(6): 927–930. Doi: 10.1111/j.1654-1103.2003.tb02228.x |
| Edgar R C, Haas B J, Clemente J C, Quince C, Knight R, 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16): 2194–2200. Doi: 10.1093/bioinformatics/btr381 |
| Edgar R C, 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19): 2460–2461. Doi: 10.1093/bioinformatics/btq461 |
| Elsabé M J, Brüchert V, Fuchs B M, 2012. Vertical shifts in the microbial community structure of organic-rich Namibian shelf sediments. African Journal of Microbiology Research, 6(17): 3887–3897. |
| Faust K, Raes J, 2012. Microbial interactions: from networks to models. Nat. Rev. Microbiol., 10(8): 538–550. Doi: 10.1038/nrmicro2832 |
| Freilich S, Kreimer A, Meilijson I, Gophna U, Sharan R, Ruppin E, 2010. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Research, 38(12): 3857–3868. Doi: 10.1093/nar/gkq118 |
| Fryer J L, Hedrick R P, 2003. Piscirickettsia salmonis: a Gramnegative intracellular bacterial pathogen of fish. J. Fish Dis., 26(5): 251–262. Doi: 10.1046/j.1365-2761.2003.00460.x |
| Fuhrman J A, 2009. Microbial community structure and its functional implications. Nature, 459(7244): 193–199. Doi: 10.1038/nature08058 |
| Guan X Y, Zhu L L, Li Y X, Xie Y X, Zhao M Z, Luo X M, 2014. Composition and variation of sediment bacterial and nirS-harboring bacterial communities at representative sites of the Bohai Gulf coastal zone, China. World J. Microb iol. Biot echnol., 30(4): 1291–1300. Doi: 10.1007/s11274-013-1553-4 |
| Hibbing M E, Fuqua C, Parsek M R, Peterson S B, 2010. Bacterial competition: surviving and thriving in the microbial jungle. Nature Reviews Microbiology, 8(1): 15–25. Doi: 10.1038/nrmicro2259 |
| Jacob M, Soltwedel T, Boetius A, Ramette A, 2013. Biogeography of deep-sea benthic bacteria at regional scale (LTER HAUSGARTEN, Fram Strait, Arctic). PLoS One, 8(9): e72779. Doi: 10.1371/journal.pone.0072779 |
| Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K, 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research, 45(D1): D353–D361. Doi: 10.1093/nar/gkw1092 |
| Korneeva V A, Pimenov N V, Krek A V, Tourova T P, Bryukhanov A L, 2015. Sulfate-reducing bacterial communities in the water column of the Gdansk Deep (Baltic Sea). Microbiology, 84(2): 268–277. Doi: 10.1134/S002626171502006X |
| Langille M G I, Zaneveld J, Caporaso J G, McDonald D, Knights D, Reyes J A, Clemente J C, Burkepile D E, Vega Thurber R L, Knight R, Beiko R G, Huttenhower C, 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotech., 31(9): 814–821. Doi: 10.1038/nbt.2676 |
| Lennon J T, Aanderud Z T, Lehmkuhl B K, Schoolmaster D R Jr, 2012. Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology, 93(8): 1867–1879. Doi: 10.1890/11-1745.1 |
| Li G X, Han X B, Yue S H, Wen G Y, Yang R M, Kusky T M, 2006. Monthly variations of water masses in the East China Seas. Cont. Shelf Res., 26(16): 1954–1970. Doi: 10.1016/j.csr.2006.06.008 |
| Li Y F, Wolanski E, Zhang H, 2015. What processes control the net currents through shallow straits? A review with application to the Bohai Strait, China. Estuar. Coast. Shelf S ci., 158: 1–11. Doi: 10.1016/j.ecss.2015.03.013 |
| Liu J W, Liu X S, Wang M, Qiao Y L, Zheng Y F, Zhang X H, 2015. Bacterial and archaeal communities in sediments of the North Chinese marginal seas. Microbial. Ecol., 70(1): 105–117. Doi: 10.1007/s00248-014-0553-8 |
| Louvado A, Gomes N C M, Simões M M Q, Almeida A, Cleary D F R, Cunha A, 2015. Polycyclic aromatic hydrocarbons in deep sea sediments: microbe-pollutant interactions in a remote environment. Sci. Total Env ron., 526: 526–312. |
| Lupatini M, Suleiman A K A, Jacques R J S, Antoniolli Z I, de Siqueira Ferreira A, Kuramae E E, Roesch L F W, 2014. Network topology reveals high connectance levels and few key microbial genera within soils. Front. Env ron. Sci., 2: 10. |
| Macalady J L, Dattagupta S, Schaperdoth I, Jones D S, Druschel G K, Eastman D, 2008. Niche differentiation among sulfur-oxidizing bacterial populations in cave waters. ISME J., 2(6): 590–601. Doi: 10.1038/ismej.2008.25 |
| Marshall S H, Gómez F A, Klose K E. 2014. The Genus Piscirickettsia. In: Rosenberg E, DeLong E F, Lory S, Stackebrandt E, Thompson F eds. The Prokaryotes: Gammaproteobacteria. Springer, Berlin Heidelberg. p. 565-573. |
| Maruyama A, Honda D, Yamamoto H, Kitamura K, Higashihara T, 2000. Phylogenetic analysis of psychrophilic bacteria isolated from the Japan Trench, including a description of the deep-sea species Psychrobacter pacificensis sp. nov. Int. J. Syst. Evol. Microbiol., 50(2): 835–846. Doi: 10.1099/00207713-50-2-835 |
| Molloy S, 2014. Environmental microbiology: disentangling syntrophy. Nature Reviews Microbiology, 12(1): 7. |
| Mu F H, Somerfield P J, Warwick R M, Zhang Z N, 2002. Large-scale spatial patterns in the community structure of benthic harpacticoid copepods in the Bohai Sea, China. The Raffles Bulletin of Zoology, 50(1): 17–26. |
| Na H, Lever M A, Kjeldsen K U, Schulz F, Jørgensen B B, 2015. Uncultured Desulfobacteraceae and crenarchaeotal group C3 incorporate 13C-acetate in coastal marine sediment. Environ. Microbiol. Rep., 7(4): 614–622. Doi: 10.1111/emi4.2015.7.issue-4 |
| Naimie C E, Blain C A, Lynch D R, 2001. Seasonal mean circulation in the Yellow Sea—a model-generated climatology. Cont. Shelf Res., 21(6-7): 667–695. Doi: 10.1016/S0278-4343(00)00102-3 |
| Nemergut D R, Schmidt S K, Fukami T, O'Neill S P, Bilinski T M, Stanish L F, Knelman J E, Darcy J L, Lynch R C, Wickey P, Ferrenberg S, 2013. Patterns and processes of microbial community assembly. Microbiology and Molecular Biology Reviews, 77(3): 342–356. Doi: 10.1128/MMBR.00051-12 |
| Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M, 1999. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research, 27(1): 29–34. Doi: 10.1093/nar/27.1.29 |
| Okuda S, Yamada T, Hamajima M, Itoh M, Katayama T, Bork P, Goto S, Kanehisa M, 2008. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Research, 36(S2): W423–W426. |
| Piontek J, Sperling M, Nöthig E M, Engel A, 2015. Multiple environmental changes induce interactive effects on bacterial degradation activity in the Arctic Ocean. Limnology and Oceanography, 60(4): 1392–1410. Doi: 10.1002/lno.10112 |
| Pontarp M, Canbäck B, Tunlid A, Lundberg P, 2012. Phylogenetic analysis suggests that habitat filtering is structuring marine bacterial communities across the globe. Microbial. Ecol., 64(1): 8–17. |
| Pujalte M J, Lucena T, Ruvira M A, Arahal D R, Macián M C. 2014. The family rhodobacteraceae. In: Rosenberg E, DeLong E F, Lory S, Stackebrandt E, Thompson F eds. The Prokaryotes: Alphaproteobacteria and Betaproteobacteria. Springer, Berlin Heidelberg. p. 439-512. |
| Sakala R M, Hayashidani H, Kato Y, Kaneuchi C, Ogawa M, 2002. Isolation and characterization of Lactococcus piscium strains from vacuum-packaged refrigerated beef. Journal of Applied Microbiology, 92(1): 173–179. Doi: 10.1046/j.1365-2672.2002.01513.x |
| Signori C N, Thomas F, Enrich-Prast A, Pollery R C G, Sievert S M, 2014. Microbial diversity and community structure across environmental gradients in Bransfield Strait, Western Antarctic Peninsula. Front. Microbiol., 5: 647. |
| Smoot M E, Ono K, Ruscheinski J, Wang P L, Ideker T, 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics, 27(3): 431–432. |
| Wang H, Wang B, Dong W W, Hu X K, 2016. Co-acclimation of bacterial communities under stresses of hydrocarbons with different structures. Sci. Rep., 6: 34588. Doi: 10.1038/srep34588 |
| Wang L P, Liu L S, Zheng B H, Zhu Y Z, Wang X, 2013. Analysis of the bacterial community in the two typical intertidal sediments of Bohai Bay, China by pyrosequencing. Mar. Pollut. Bull., 72(1): 181–187. Doi: 10.1016/j.marpolbul.2013.04.005 |
| Wang L P, Zheng B H, Lei K, 2015. Diversity and distribution of bacterial community in the coastal sediments of Bohai Bay, China. Acta Oceanologica Sinica, 34(10): 122–131. Doi: 10.1007/s13131-015-0719-3 |
| Wang L P, Zheng B H, Nan B X, Hu P L, 2014. Diversity of bacterial community and detection of nirS-and nirK- encoding denitrifying bacteria in sandy intertidal sediments along Laizhou Bay of Bohai Sea, China. Mar. Pollut. Bull., 88(1-2): 215–223. Doi: 10.1016/j.marpolbul.2014.09.002 |
| Wu H B, Guo Y T, Wang G H, Dai S K, Li X, 2011. Composition of bacterial communities in deep-sea sediments from the South China Sea, the Andaman Sea and the Indian Ocean. African Journal of Microbiology Research, 5(29): 5273–5283. |
| Zhang J J, Kobert K, Flouri T, Stamatakis A, 2014. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics, 30(5): 614–620. Doi: 10.1093/bioinformatics/btt593 |
| Zhang J, Yu Z G, Raabe T, Liu S M, Starke A, Zou L, Gao H W, Brockmann U, 2004. Dynamics of inorganic nutrient species in the Bohai seawaters. J. Mar. Syst., 44(3-4): 189–212. Doi: 10.1016/j.jmarsys.2003.09.010 |
| Zhang X M, Liu W, Schloter M, Zhang G M, Chen Q S, Huang J H, Li L H, Elser J J, Han X G, 2013. Response of the abundance of key soil microbial nitrogen-cycling genes to multi-factorial global changes. PLoS One, 8(10): e76500. Doi: 10.1371/journal.pone.0076500 |
| Zheng B H, Wang L P, Liu L S, 2014. Bacterial community structure and its regulating factors in the intertidal sediment along the Liaodong Bay of Bohai Sea, China. Microbiol. Res., 169(7-8): 585–592. Doi: 10.1016/j.micres.2013.09.019 |