 2018, Vol. 36
2018, Vol. 36Institute of Oceanology, Chinese Academy of Sciences
Article Information
- YU Haiyang(俞海洋), DU Xinxin(杜昕昕), LI Xiaojing(李晓静), QU Jiangbo(曲江波), ZHU He(朱鹤), ZHANG Quanqi(张全启), WANG Xubo(王旭波)
- Genome-wide identification and transcriptome-based expression analysis of sox gene family in the Japanese flounder Paralichthys olivaceus
- Chinese Journal of Oceanology and Limnology, 36(5): 1731-1745
- http://dx.doi.org/10.1007/s00343-018-7216-4
Article History
- Received Jul. 13, 2017
- accepted in principle Aug. 17, 2017
- accepted for publication Sep. 11, 2017




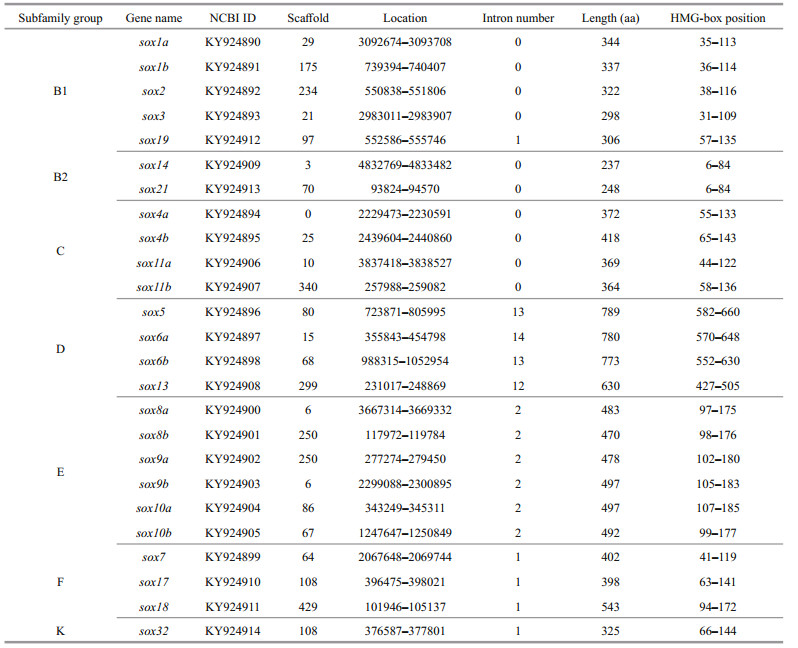
Sox genes, characterized by the conserved high mobility group (HMG) box, encode a class of transcription factors with high sequence similarity to sex determining region of Y chromosome (SRY) gene in animals. Since 1990, when the first Sry gene was identified in human and mouse, more than 100 sox genes have been discovered in mammals, birds, reptiles, fishes, and insects (Sinclair et al., 1990; Gao et al., 2016). According to the conservatism of protein and nucleic acid sequences, sox genes are named as sox1 to sox32, and classified as 12 subfamilies (A, B1, B2, C, D, E, F, G, H, I, J, and K).
With the development of genome-wide sequencing, an increasing number of sox genes were identified from different species, for instance, five in nematode (Caenorhabditis elegans) (C. elegans Sequencing Consortium, 1998), seven in calcareous sponge (Sycon ciliatum) (Fortunato et al., 2012), eight in fruitfly (Drosophila melanogaster) (Crémazy et al., 2001) and African malaria mosquito (Anopheles gambiae) (Wilson and Dearden, 2008), nine in honey bee (Apis mellifera), red flour beetle (Tribolium castaneum) and jewel wasp (Nasonia vitripennis) (Wilson and Dearden, 2008), 14 in starlet sea anemone (Nematostella vectensis) (Magie et al., 2005), 19 in Japanese medaka (Oryzias latipes) (Cui et al., 2011), 20 in mouse (Mus musculus) and human (Homo sapiens) (Schepers et al., 2002), 23 in tongue sole (Cynoglossus semilaevis) (Gao et al., 2016), 24 in torafugu (Takifugu rubripes) (Koopman et al., 2004), and 27 in Nile tilapia (Oreochromis niloticus) (Wei et al., 2016) (Table 1). Genome-wide comparison has provided valuable information to understand the function and evolution of sox genes. With the advancing knowledge on sox gene family, researchers have discovered the essential function of sox genes as important transcription factors in the regulation of diverse growth and development processes. For instance, sox gene knockout and mutation have revealed that the function of sox genes in many aspects, including chondrogenesis (Ng et al., 1997), neurogenesis (Wegner, 2011), early embryonic development (Kikuchi et al., 2001), hematopoiesis (Chung et al., 2010), stemness (Tanimura et al., 2013), angiogenesis (Pennisi et al., 2000; Downes and Koopman, 2001; Matsui et al., 2006), cardiogenesis (Zhang et al., 2005), hair development (Irrthum et al., 2003), and sex determination and differentiation (Foster et al., 1994; Vidal et al., 2001). Specifically, sox9 has been confirmed to be an essential factor for the proper proliferation and survival of medaka germ cells (Nakamura et al., 2012). Sox7 and sox18 play redundant roles in vascular development and arteriovenous specification in zebrafish (Cermenati et al., 2008; Herpers et al., 2008). Sox17 has unique effects on primitive erythropoiesis in zebrafish (Chung et al., 2010).

Japanese flounder (Paralichthys olivaceus) is a valuable economic teleost in China, Japan, and Korea. Recent developments in genome and transcriptome sequencing techniques provide a better approach to identify Japanese flounder sox genes at a genome scale level. In the present study, we used the genome and transcriptome sequencing to identify sox genes in this species. Gene structure and expression profiles of sox genes were also analyzed. Our results provided a better way to further investigate the evolution and function of sox gene family in Japanese flounder.
2 MATERIAL AND METHOD 2.1 Ethics statementThe Japanese flounder individuals used in this study were obtained from Yellow Sea Aquatic Product Co. Ltd., Yantai, Shandong, China. All experimental procedures and investigations were supervised and approved by Ocean University of China, and were performed in accordance with the guidelines of China Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training (State science and technology commission of the People's Republic of China for No. 2, November 14, 1988).
2.2 Fish and sample collectionJapanese flounder individuals (three females and three males) of one year old were chosen from a larger cohort population. The fish were anesthetized with MS-222 (30 mg/mL) and killed by severing the spinal cord. Tissue samples, including heart, liver, spleen, kidney, brain, gill, muscle, intestines, stomach, testis, and ovary, were collected respectively frozen immediately in liquid nitrogen, and then stored in -80℃ for RNA extraction.
Embryos of different development stages were collected in the breeding season (late April). In order to facilitate sampling, we divided the embryo development process of Japanese flounder into six stages, namely, Stage 1 (from two cells to morula), Stage 2 (from early gastrula to late somites), Stage 3 (from hatching to 2 d after hatching), Stage 4 (before metamorphosis), Stage 5 (metamorphosis stage 1 to 2), and Stage 6 (metamorphosis stage 3 to 5). Samples of different stages were collected respectively and stored in -80℃ for RNA extraction.
2.3 RNA extraction and illumina sequencingTotal RNA was extracted from tissue and development stage samples using Trizol reagent (Invitrogen, Carlsbad CA, USA) according to the manufacturer's protocol. DNA contamination was removed by DNaseI (TaKaRa, Dalian, China). The high-quality RNA from each sample was used to construct the llumina sequencing libraries by Illumina TruSeq mRNA Stranded Sample Preparation Kit (Illumina, San Diego CA, USA) according to the manufacturer's protocol and a previous study (Zhang et al., 2016). All constructed cDNA library were quenched by Beijing Genomics Institute (BGI, Shenzhen, China). Base on the acquired sequence reads, the fragments per kilobase of exon model per million mapped reads (FPKM) was calculated to measure gene expression levels (Garber et al., 2011; Trapnell et al., 2012) according to the formula as follows (Weitschek et al., 2015):

where Nf is the count of mapped fragments, F is the total number of the mapped fragments, and L is the length (base pairs) of all exons of sox gene.
2.4 Identification of the Sox genesIn order to perform the analysis in a more comprehensive way, two procedures were used to identify the candidate sox genes in Japanese flounder genome and transcriptome. Sox protein sequences of human (Homo sapiens), mouse (Mus musculus), zebrafish (Danio rerio), fugu (Takifugu rubripes), and tilapia (Oreochromis niloticus) were retrieved from Ensemble (http://asia.ensembl.org/index.html) and NCBI (https://www.ncbi.nlm.nih.gov/) databases. All these Sox protein-coding sequences were queried against the Japanese flounder genome (Zhang, 2016, unpublished data) and transcriptome (Zhang, 2016, unpublished data) by local TBlastx search with the threshold level of E-value of 1e-5 (Altschul et al., 1997). Meanwhile, the conserved HMG box sequence of vertebrate Sox proteins (DHVKRPMNAFMVWS-RGERRKIAQQNPDMHNSEISKRLGKRWKLLS-ESEKRPFIEEAERLRAQHMKDYPDYKYRPRR-KKK) (Wang et al., 2002) was used as a query sequence in local TBlastn search against the Japanese flounder genome and transcriptome with the threshold level of E-value of 1e-5. The results of these two procedures were integrated together, and the Blast search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and phylogenetic analysis were used to assess the accuracy of the candidate sox genes. Besides, the exon-intron structure of Japanese flounder sox genes were mapped by Blastn program.
2.5 Phylogenetic analysis of Sox geneBefore phylogenetic analysis, the Sox protein sequences of medaka (Oryzias latipes), spotted gar (Lepisosteus oculatus), and fruit fly (Drosophila melanogaster) were retrieved from Ensemble and NCBI. Sox proteins sequences of nine species (human, mouse, fruit fly, fugu, zebrafish, tilapia, medaka, spotted gar, and Japanese flounder) were used to construct the phylogenetic tree. The accession numbers of these protein were available in Table S1. Multiple Sequence alignment of all the Sox protein sequences were implemented by ClustalW before evolutionary tree analysis. The phylogenetic tree was constructed by the neighbor-joining method with Poisson model implemented in MEGA 7.0 program with bootstrap of 10 000 replications. Specifically, the evolutionary distances were computed using the Poisson correction method and all positions containing gaps and missing data were eliminated (Zuckerkandl and Pauling, 1965; Saitou and Nei, 1987; Kumar et al., 2016).
2.6 Analysis of Sox gene expression base on transcriptomeThe transcriptomes of eleven tissues (heart, liver, spleen, kidney, brain, gill, muscle, intestines, stomach, testis, and ovary) and six embryonic development stages (Stage 1–6) were used to analyze sox gene expression in Japanese flounder. The sox gene expression levels in different tissues and stages were evaluated by normalized FPKM value. For sox gene expression in different tissues and embryonic development stages, FPKM≥1 was considered reasonable, and FPKM≥10 was considered as a threshold for high expression (Hart et al., 2013; Tsagaratou et al., 2014).
The biased/specific expression of sox genes in gonads (testis or ovary) was also analyzed based on RNA-Seq data. The testis/ovary-biased expressed candidate genes were identified when "FPKM≥1" and "|log2(FPKMovary/FPKMtestis)|≥2". The testis/ovary-specific expressed genes were identified when "FPKM≥3" in one gonads, but "FPKM≤1" in the other.
2.7 Sox gene expression quantified by qPCRExtracted total RNA was transcribed by M-MLV Reverse Transcriptase (TaKaRa) enzyme according to the manufacturer's protocol. The primers used in this experiment were designed by Primer Primer 5.0 and listed in Table S2. Pre-experiment was conducted to test the specificity of primers. qPCR was performed in Light-Cycler 480 (Roche, Forrentrasse, Switzerland) with in 20 μL reaction volume, which contained cDNA templates 10 ng, primers (FW/RV) and 2×SYBR Green qPCR Master Mix (US Everbright Inc.). β-actin was selected as a reference gene (Zhang et al., 2013). The reaction procedure consisted of an initial polymerase activation of 5 min at 94℃, followed by 40 cycles at 94℃ (15 s) and 60℃ (45 s). The data were analyzed by the 2-ΔΔCt method. This experiment was performed according to a previous study (Gao et al., 2015).
3 RESULT 3.1 Sox gene subfamilies in Japanese flounderBased on the relatively conserved HMG box domain of vertebrate Sox proteins (Bowles et al., 2000), the conserved HMG-box domain of vertebrate and Sox protein-coding sequences of five species were used to search against the Japanese flounder genome and transcriptome by Local Alignment Search Tool (BLAST+ 2.6.0). A total of 25 sox genes were isolated and identified from Japanese flounder genome which could be divided into seven subfamilies (Table 2), that is, subfamily B1 (including Posox1a, Posox1b, Posox2, Posox3, and Posox19), subfamily B2 (including Posox14 and Posox21), subfamily C (including Posox4a, Posox4b, Posox11a, and Posox11b), subfamily D (including Posox5, Posox6a, Posox6b, and Posox13), subfamily E (including Posox8a, Posox8b, Posox9a, Posox9b, Posox10a, and Posox10b), subfamily F (including Posox7, Posox17, and Posox18), subfamily K (including Posox32). Compare with human and mouse, which had only one sox gene copy, some teleost sox genes had two duplicates (except spotted gar (Hermansen et al., 2016)) (Table 3). These results suggested that these novel sox isoforms might derive from a teleost-specific evolutionary process.
The 25 Sox protein sequences identified from Japanese flounder were used to perform online protein sequence analysis by SMART (http://smart.embl-heidelberg.de/). The results showed that all the Sox proteins had a conserved HMG box of 79 amino acid residues (Gubbay et al., 1990). As shown in Fig. 1, both the motif sequence (positions 5-10) and the extended motif sequence (position 5-13) were highly conserved for all Sox sequences except PoSox32. Besides, some other fragments in the HMG box were also highly conserved. Interestingly, similar to Sox32 in medaka (Cui et al., 2011), and tongue sole (Gao et al., 2016), Japanese flounder Sox32 could be distinguished from other Sox proteins. For example, the amino acid at position 7 in PoSox32 motif was L, but it was M in the other Sox sequences. Moreover, residues at positions 11-13 in PoSox32 HMG box were IIW, which were also different from the highly conserved MVW of other Sox proteins in these positions. These results indicated that the function and evolution of Sox32 in teleosts might be more complex compared with other vertebrates.

|
| Figure 1 Conserved HMG box domains of Japanese flounder Sox proteins All HMG box domains were predicted by SMART online program (http://smart.embl-heidelberg.de/). ClustalW and GENEDOC program were used to perform multiple sequence alignment of the amino acid sequence. B1, B2, C, D, E, F and K indicate the seven subfamilies of Japanese flounder Sox proteins. Residues in dark are conserved among all the sequences, and residues in gray are conserved in most sequences. |
The exon-intron structure of Japanese flounder sox genes were drawn by online software GSDS 2.0 (Gene Structure Display Server, http://gsds.cbi.pku.edu.cn/) (Hu et al., 2015). As shown in Fig. 2, the structure of Japanese flounder sox genes were diverse, which could be summarized as four categories—no intron, one intron, two introns, and multiple introns. With except Posox19, all sox genes in Subfamilies B1, B2, and C belonged to "no intron", which meant no intron was found in these genes. Subfamilies F and K belonged to "one intron", possessing only one intron in the sox gene structure. Similarly, all sox genes in subfamily E (belonging to "two introns") had two introns, and all sox genes of subfamily D had more than two introns, belonging to "multiple introns". It is worth noting that the number of introns in subfamily D was much more than that of the other subfamilies, which might be caused by its special evolutionary processes. We assumed that the diverse genomic organization of sox genes might generate from an early divergence of the different genes during evolution (Roose et al., 1999).

|
| Figure 2 Structure of Japanese flounder sox genes Online software GSDS 2.0 (http://gsds.cbi.pku.edu.cn/) was used to draw sox genes structure. Rectangle and double slash represent exon and omitted sequences, respectively. The HMG box domains are marked gray. B1, B2, C, D, E, F and K indicate the seven subfamilies of Japanese flounder sox genes. |
As shown in Fig. 2, the sox genes from the same subfamily had similar or the same exon-intron structure. This finding was consistent with former studies. For instance, no intron in the HMG box has been reported in Subfamilies A, B, C, and G in vertebrate sox genes. However, seven of the eight sox genes, members of Subfamilies B and C, in nematode (Caenorhabditis elegans) and fruitfly (Drosophila melanogaster) have introns in their HMG boxes (Bowles et al., 2000). These results indicated these introns in Subfamilies B and C have been lost during deuterostome evolutionary process. Besides, the introns of Subfamilies D, E and F were relatively conserved in fruitfly and vertebrates, which suggested that they were ancient introns existing before vertebrates appeared. The positions of introns are highly conserved, and rarely changed in orthologues (Kersanach et al., 1994). Therefore, the gain and loss of intron in sox genes might indicate that a series of genetic rearrangements had happened during evolutionary process.
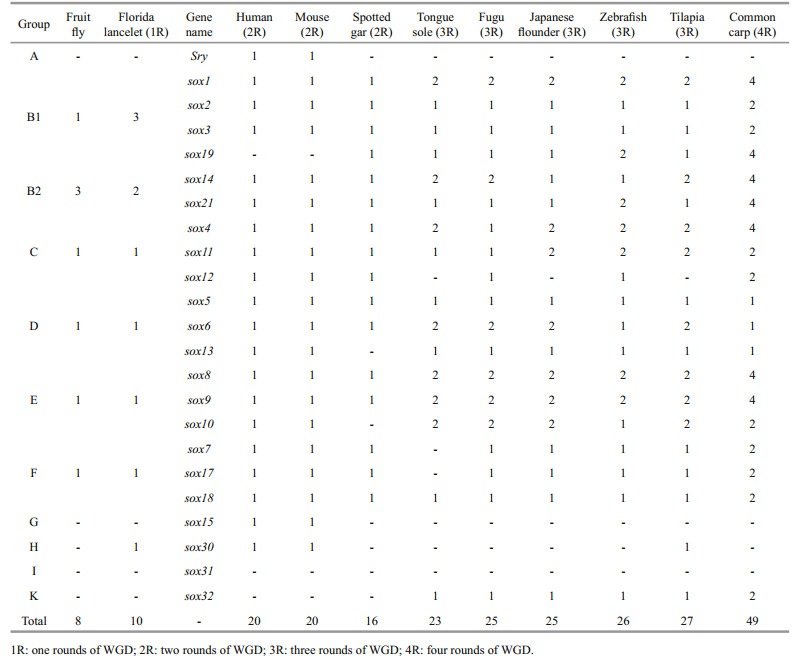
3.4 Sox gene evolution in Japanese flounderThe Sox protein sequences of human, mouse, fruit fly, fugu, zebrafish, tilapia, medaka, spotted gar, and Japanese flounder were used to construct the phylogenetic tree by MEGA 7.0 program. As shown in Fig. 3, the sox genes used in the analysis could be divided into ten subfamilies (including A, B1, B2, C, D, E, F, G, H, and K), and high degree of consistency was found among different subfamilies. Notably, a closer evolutionary relationship was detected between subfamily K and subfamily F, subfamily B1 and subfamily B2, and subfamily E and subfamily H. Interestingly, subfamilies A, K, H, and G had only one member. Moreover, Sry in subfamily A and sox15 in subfamily G were found exclusively in human and mouse, while sox32 in subfamily K was found exclusively in teleosts. We speculated that the generation or loss of some sox genes in teleosts might result from teleost-specific whole-genome duplication (WGD) (Chung et al., 2011).

|
| Figure 3 Phylogenetic tree of sox genes of Japanese flounder and other animals The tree was constructed by MEGA 7.0 program using neighbor-joining methods. Dm: Drosophila melanogaster; Dr: Danio rerio; Hs: Homo sapiens; Lo: Lepisosteus oculatus; Mm: Mus musculus; Ol: Oryzias latipes; On: Oreochromis niloticus; Po: Paralichthys olivaceus; Tr: Takifugu rubripes. A, B1, B2, C, D, E, F, G, H and K indicate the ten sox subfamilies. |
It was interesting to note that the majority of sox genes could be clustered into their respective subfamilies. This phenomenon provided a strong evidence supporting that the same subfamily might have a common evolutionary origin. However, there were also some abnormalities in the phylogenetic tree. For instance, members of subfamily A (HsSRY and MmSRY) were not able to be clustered into one group, suggesting that subfamily A might not be robustly monophyletic. Bowles et al. also encountered the same problem in their study (Bowles et al., 2000), and thought that the aberrant behavior of SRY (members of Group A do not form a monophyletic group) in the particular phylogenetic analysis is likely related to its remarkably high evolutionary rate. This unexpected result might reflect that the sox genes were still at a relatively rapid rate of divergence.
Following the first two rounds of WGD, the third WGD event has shaped the teleost evolution, and generated the most diverse vertebrate group, providing abundant raw materials for evolutionary adaptation and innovation (Glasauer and Neuhauss, 2014). After WGD, orthologues have different fates, such as, subfunctionalization, neofunctionalization, and dosageselection (Force et al., 1999). The different number of sox gene among species might be varied with different rounds of genome duplication (Table 3), for example, eight sox genes in fruit fly (Crémazy et al., 2001), 10 in florida lancelet (one rounds of WGD, 1R), 20 in human (two rounds of WGD, 2R), 20 in mouse (2R), 16 in spotted gar (2R), 23 in tongue sole (three rounds of WGD, 3R) (Gao et al., 2016), 25 in fugu (3R), 25 in Japanese flounder, 26 in zebrafish (3R), 27 in tilapia (3R) (Wei et al., 2016), and 49 in common carp (four rounds of WGD, 4R). Together with previous reports, it was reasonable to deduce that sox gene family might also undergo an explosive growth along with the process of WGD.
3.5 Expression profiles of Japanese flounder Sox genesFPKM value was used to draw the heat map of sox gene expression in Japanese flounder (Fig. 4). FPKM≥1 was considered as a reasonable expression level and FPKM≥10 was considered as a high expression level. Results showed that Japanese flounder sox4b, sox9a, and sox9b had higher levels, while the other sox genes had very low or negligible expression levels in gill. Interestingly, most sox genes (except sox6a, sox6b, sox8a, sox11b, and sox32) had relatively higher levels in brain, and sox1a, sox1b, sox3, sox4a, sox10a, sox10b, sox13, sox14, sox17, and sox19 had the strongest expression in brain compared with their expression in other tissues. Moreover, expression levels of sox9a in heart, sox6b in muscle, sox7 in intestines, and sox7 and sox11a in ovary were all relatively higher. Especially, sox8b expression level was extremely high in testis, which might imply its involvement in the development of Japanese flounder testis. We also noticed the weak expression levels of sox6a, sox11b, and sox32, whose role in Japanese flounder development needed further investigation.

|
| Figure 4 Spatial expression profiles of Japanese flounder sox genes in tissues Each row represent a sox gene, and each column represents a sample. Each cell in the heat map corresponds to an expression level, with light blue for underexpression, and dark blue for overexpression (see the color scale). The number in cell are FPKM values. |
Furthermore, the expression of sox genes in six embryonic development stages of Japanese flounder was also analyzed (Fig. 5). The results revealed that sox genes had special temporal expression patterns during embryonic development. Most sox genes, especially sox3, sox4b, sox11b, and sox19 were highly expressed in stages 1–4 (from two cells to before metamorphosis), while their levels gradually decreased in Stage 5 and Stage 6. We conjectured that this phenomenon was related to the change in sox gene function in embryogenesis, neurogenesis, oligodendrocyte development, chondrogenesis, and neural crest cell development, amongst others (Jiang et al., 2013). Similarly, previous studies have reported that sox genes were widely and dynamically expressed in various phases of embryogenesis (Kamachi et al., 2000; Sánchez-Soriano and Russell, 2000). Besides, we also noticed that sox5, sox6a, and sox8a had lower levels in all six stages.

|
| Figure 5 Temporal expression profiles of Japanese flounder sox genes during embryonic development Each row represent a sox gene, and each column represents a stage. Each cell in the heat map corresponds to an expression level, with light blue for underexpression, and dark blue for overexpression (see the color scale). The number in cell are FPKM values. Stage 1 (from two cells to morula); stage 2 (from early gastrula to late somites); stage 3 (from hatching stage to 2 d after hatching); stage 4 (before metamorphosis); stage 5 (metamorphosis stages 1 to 2); stage 6 (metamorphosis stages 3 to 5). |
Members of sox gene family were first identified as testis determining genes and considered to be related to gender differentiation and gonadal development (Nagai, 2001). Japanese flounder is an important economic fish in China, but its sex determination mechanisms have not been elucidated until now.
In our study, FPKM data were used to analyze gonad-biased/specific sox genes. These results showed that sox2 and sox7 had biased expression in ovary, and sox8b in testis (Fig. 6); moreover, sox8a had specific expression in testis, and sox10a in ovary (Fig. 4). Sox9, which has been considered as a sex-related gene in mammal, was also verified by qPCR. Consistent results were found in qPCR verification (Fig. 7). Resultsed showed that sox2, sox7, sox10a, and sox10b were predominantly expressed in ovary, and sox8a, sox8b, sox9a, and sox9b were mainly expressed in testis.

|
| Figure 6 Gonad-biased expression of Japanese flounder sox genes |

|
| Figure 7 Relative expression of sox2, sox7, sox8a, sox8b, sox9a, sox9b, sox10a and sox10b in Japanese flounder testis and ovary examined by qPCR (n=3) β-actin was used as an internal reference (** P < 0.01). |
This study identified 25 sox genes from Japanese flounder genome and transcriptome. These sox genes could be divided into seven subfamilies (B1, B2, C, D, E, F, and K). During the evolution history of the sox gene family, the number of sox gene increased significantly. Ever since 1990, when Andrew H. Sinclair, et al (Sinclair et al., 1990) discovered a new transcribed gene Sry in human, more than 12 sox subfamilies have been discovered in vertebrates and invertebrates. The subfamilies in invertebrates like nematode (Caenorhabditis elegans) and fruit fly (Drosophila melanogaster) have only one member (C. elegans Sequencing Consortium, 1998; Crémazy et al., 2001), and generate multiple members in early vertebrate evolution. Some members of these subfamilies in teleosts have two parallel orthologous genes, for instance sox1a and sox1b, and especially in Group K, a newly discovered subfamily, which was found exclusively in teleosts. These results implied that teleosts had experienced a specific genome duplication after splitting from the lineage that evolved into human (Amores et al., 2011).
4.2 The evolution and duplication of Sox genes in teleostsThe results of this study also validated the hypothesis that sox genes of teleosts might have undergone expansion during the third rounds of WGD. On the one hand, most sox genes, such as Posox1a/1b and Posox8a/8b, possessed two duplicates in teleosts. On the other hand, sox32 in subfamily K was found exclusively in teleosts. Studies in zebrafish showed that sox32 was a key regulator of endoderm formation (Shin et al., 2008), suggesting that teleost sox32 was an indispensable factor for endodermal differentiation. Intriguingly, sox30 was considered to exist only in mammals, and consistently in our study, sox30 in subfamily H could only found in human and mouse (except for tilapia), but absent in spotted gar, medaka, tongue sole, fugu, Japanese flounder, zebrafish, and common carp. Studies in tilapia and mouse showed that sox30 was expressed exclusively in gonads, suggesting that sox30 was probably a gonad-specific gene (Fei et al., 2010). Besides, this study also suggested that sox genes had undergone an expansion following teleost genome duplication (Fei et al., 2010; Wei et al., 2016). All these results supported that teleost experienced a specific third WGD, and that the evolution and functions of sox30 and sox32 might be more complex, which needed further verification.
4.3 The Sox genes related to Japanese flounder neurogenesisA previous research has demonstrated that sox genes, as transcription factors, are involved in the decision of various important cell fates during development (Jay et al., 1997). Intriguingly, in our study, sox1a, sox1b, sox3, sox13, sox14, and sox19 had specific expression in Japanese flounder brain (Fig. 4). Moreover, the highest level of sox3 was detected in brain (FPKM≥10). Existed research has shown that sox1, sox2, and sox3 are critical determinants of neurogenesis, which can keep neural cells undifferentiated by counteracting with proneural proteins (Bylund et al., 2003). Studies in mouse showed that sox13 was expressed in the developing central nervous system (CNS), suggesting its significance in neurogenesis (Wang et al., 2006). Although the role of sox14 during neural development remains unclear, some studies have suggested its implications in neural development (Popovic et al., 2014). In zebrafish, sox19 was considered to be the earliest molecular marker of CNS (Vriz et al., 1996). Combined with these points of view, we inferred that the six sox genes (sox1a, sox1b, sox3, sox13, sox14, and sox19) might have an important function in Japanese flounder neurogenesis.
4.4 The Sox genes related to Japanese flounder gonad developmentGonad-biased sox genes (sox2, sox7, and sox8b) and gonad-specific sox genes (sox8a and sox10a) of Japanese flounder were also discovered in this study. Previous research showed that sox8 and sox9, especially the latter, were highly expressed during mammalian testis development. In mouse, sox9 mutations can cause gender reversal or severe infertility, and sox8 mutations can lead to a decline in fertility. Besides, mouse sox9 and sox8 function at earlier and later stages of testis development, respectively (Barrionuevo and Scherer, 2010). Consistently, Japanese flounder sox8a was specifically expressed in testis, sox8b had extremely high expression in testis, whereas sox9 had moderate expression in testis and ovary, which might imply the functional differentiation between sox8 and sox9.
In vitro transfection assays showed that sox10a might be involved in the regulation of cyp19a1a gene in orange-spotted grouper (Epinephelus coioides) (Liu et al., 2012). Besides, cyp19 was detected to be mainly expressed in Japanese flounder ovary (Luckenbach et al., 2005). Combined with these findings, we speculated that the function of Japanese flounder sox10a, specifically expressed in ovary, might also involve the regulation of cyp19a1a gene. Interestingly, sox2 in chick and sox7 in zebrafish were considered to be related to neurogenesis and vascular development, respectively (Bylund et al., 2003; Herpers et al., 2008), but in our study, Japanese flounder sox2 and sox7 were ovary-biased expressed genes, implying that sox2 and sox7 might possess specific functions in Japanese flounder ovary development.
4.5 Multiple functions of Sox genes in vertebrateIn addition, multiple functions have been found in other sox genes. For example, sox5, sox6, sox17, and sox30 were discovered in mouse testis, and considered to be involved in spermatogonial differentiation and spermatogenesis (Kanai et al., 1996; Wunderle et al., 1996; Ohe et al., 2009; Han et al., 2014). Functional experiments further indicated that mouse sox15 played an important role in developing testis (Sarraj et al., 2003). Mouse sox3 was important for oocyte development, testis differentiation and gametogenesis, and sox4 might play an integral role in CNS development. Orange-spotted grouper (Epinephelus coioides) sox11b was decreased significantly during sex change, indicating that sox11b might be involved in oogenesis and sex change process (Zhang et al., 2008). Rainbow trout (Oncorhynchus mykiss) sox24 played roles during oogenesis (Kanda et al., 1998). Nevertheless, the function of Japanese flounder sox genes is still a blank area, which needs further extensive research.
5 CONCLUSIONIn this study, 25 sox genes were identified from Japanese flounder genome and transcriptome. Through gene structure, phylogenetic and expression analyses, the conserved structure and various expression patterns of sox genes were uncovered. We also discovered gonadal-biased and gonadal-specific expression of some Japanese flounder sox genes. This study would establish the foundation for further sox gene function analysis in Japanese flounder.
6 DATA AVAILABILITY STATEMENTThe sequences of sox genes in Japanese flounder are available from GenBank under the accessions KY924890–KY924914. The accession numbers of other species' sox genes used in this article are provided in Supplementary material file. Additional supporting data can acquire from the corresponding author upon reasonable request.
7 CONFLICT OF INTEREST STATEMENTWe declare no conflict of interest.
Electronic supplementary materialSupplementary material (Supplementary Fig.1 and Tables S1–S2) is available in the online version of this article at https://doi.org/10.1007/s00343-018-7216-4.
Altschul S F, Madden T L, Schäffer A A, Zhang J H, Zhang Z, Miller W, Lipman D J. 1997. Gapped BLAST and PSIBLAST:a new generation of protein database search programs. Nucleic Acids Research, 25(17): 3 389-3 402.
DOI:10.1093/nar/25.17.3389 |
Amores A, Catchen J, Ferrara A, Fontenot Q, Postlethwait J H. 2011. Genome evolution and meiotic maps by massively parallel DNA sequencing:spotted gar, an outgroup for the teleost genome duplication. Genetics, 188(4): 799-808.
DOI:10.1534/genetics.111.127324 |
Barrionuevo F, Scherer G. 2010. SOX E genes:SOX9 and SOX8 in mammalian testis development. The International Journal of Biochemistry & Cell Biology, 42(3): 433-436.
|
Bowles J, Schepers G, Koopman P. 2000. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Developmental Biology, 227(2): 239-255.
DOI:10.1006/dbio.2000.9883 |
Bylund M, Andersson E, Novitch B G, Muhr J. 2003. Vertebrate neurogenesis is counteracted by Sox1-3 activity. Nature Neuroscience, 6(11): 1 162-1 168.
DOI:10.1038/nn1131 |
elegans Sequencing Consortium C.. 1998. Genome sequence of the nematode C. elegans:a platform for investigating biology. Science, 282(5396): 2 012-2 018.
DOI:10.1126/science.282.5396.2012 |
Cermenati S, Moleri S, Cimbro S, Corti P, Del Giacco L, Amodeo R, Dejana E, Koopman P, Cotelli F, Beltrame M. 2008. Sox18 and Sox7 play redundant roles in vascular development. Blood, 111(5): 2 657-2 666.
DOI:10.1182/blood-2007-07-100412 |
Chung I, Leung A, Ma C, Fung T. 2010. The role of Sox genes(Group F) in zebrafish hematopoiesis. Experimental Hematology, 38(9S): S71-S72.
|
Chung M I S, Ma A C H, Fung T K, Leung A Y H. 2011. Characterization of Sry-related HMG box group F genes in zebrafish hematopoiesis. Experimental Hematology, 39(10): 986-998.
DOI:10.1016/j.exphem.2011.06.010 |
Crémazy F, Berta P, Girard F. 2001. Genome-wide analysis of Sox genes in Drosophila melanogaster. Mechanisms of Development, 109(2): 371-375.
|
Cui J, Shen X, Zhao H, Nagahama Y. 2011. Genome-wide analysis of Sox genes in Medaka (Oryzias latipes) and their expression pattern in embryonic development. Cytogenetic and genome research, 134(4): 283-294.
DOI:10.1159/000329480 |
Downes M, Koopman P. 2001. SOX18 and the transcriptional regulation of blood vessel development. Trends in Cardiovascular Medicine, 11(8): 318-324.
DOI:10.1016/S1050-1738(01)00131-1 |
Fei H, Wang Z J, Wu F R, Liu Z H, Huang B F, Wang D S. 2010. Characterization, phylogeny, alternative splicing and expression of Sox30 gene. BMC Molecular Biology, 11: 98.
DOI:10.1186/1471-2199-11-98 |
Force A, Lynch M, Pickett F B, Amores A, Yan Y L, Postlethwait J. 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics, 151(4): 1 531-1 545.
|
Fortunato S, Adamski M, Bergum B, Guder C, Jordal S, Leininger S, Zwafink C, Rapp H, Adamska M. 2012. Genome-wide analysis of the sox family in the calcareous sponge Sycon ciliatum:multiple genes with unique expression patterns. EvoDevo, 3(1): 14.
DOI:10.1186/2041-9139-3-14 |
Foster J W, Dominguez-Steglich M A, Guioli S, Kwok C, Weller P A, Stevanović M, Weissenbach J, Mansour S, Young I D, Goodfellow P N, Brook J D, Schafer A J. 1994. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature, 372(6506): 525-530.
DOI:10.1038/372525a0 |
Gao J N, Li P Z, Zhang W, Wang Z G, Wang X B, Zhang Q Q. 2015. Molecular cloning, promoter analysis and expression profiles of the sox3 gene in Japanese Flounder, Paralichthys olivaceus. International Journal of Molecular Sciences, 16(11): 27 931-27 944.
DOI:10.3390/ijms161126079 |
Gao J, Ma J L, Liu Y, Shao C W, Jia X D, Chen S L. 2016. Bioinformatics analysis of Sox gene family in Cynoglossus semilaevis. Progress in Fishery Sciences, 37(2): 41-48.
(in Chinese with English abstract) |
Garber M, Grabherr M G, Guttman M, Trapnell C. 2011. Computational methods for transcriptome annotation and quantification using RNA-seq. Nature Methods, 8(6): 469-477.
DOI:10.1038/nmeth.1613 |
Glasauer S M K, Neuhauss S C F. 2014. Whole-genome duplication in teleost fishes and its evolutionary consequences. Molecular Genetics and Genomics, 289(6): 1 045-1 060.
DOI:10.1007/s00438-014-0889-2 |
Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Münsterberg A, Vivian N, Goodfellow P, Lovell-Badge R. 1990. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature, 346(6281): 245-250.
DOI:10.1038/346245a0 |
Han F, Dong Y, Liu W B, Ma X X, Shi R H, Chen H Q, Cui Z H, Ao L, Zhang H D, Cao J, Liu J Y, Lobaccaro J M A. 2014. Epigenetic regulation of Sox30 is associated with testis development in mice. PLoS One, 9(5): e97203.
DOI:10.1371/journal.pone.0097203 |
Hart T, Komori H K, LaMere S, Podshivalova K, Salomon D R. 2013. Finding the active genes in deep RNA-seq gene expression studies. BMC Genomics, 14(1): 778.
DOI:10.1186/1471-2164-14-778 |
Hermansen R A, Hvidsten T R, Sandve S R, Liberles D A. 2016. Extracting functional trends from whole genome duplication events using comparative genomics. Biological Procedures Online, 18(1): 11.
DOI:10.1186/s12575-016-0041-2 |
Herpers R, van de Kamp E, Duckers H J, Schulte-Merker S. 2008. Redundant roles for Sox7 and Sox18 in arteriovenous specification in zebrafish. Circulation Research, 102(1): 12-15.
DOI:10.1161/CIRCRESAHA.107.166066 |
Hu B, Jin J, Guo A Y, Zhang H, Luo J, Gao G. 2015. GSDS 2.0:an upgraded gene feature visualization server. Bioinformatics, 31(8): 1 296-1 297.
DOI:10.1093/bioinformatics/btu817 |
Irrthum A, Devriendt K, Chitayat D, Matthijs G, Glade C, Steijlen P M, Fryns J P, Van Steensel M A, Vikkula M. 2003. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. The American Journal of Human Genetics, 72(6): 1 470-1 478.
DOI:10.1086/375614 |
Jay P, Sahly I, Gozé C, Taviaux S, Poulat F, Couly G, Abitbol M, Berta P. 1997. SOX22 is a new member of the SOX gene family, mainly expressed in human nervous tissue. Human Molecular Genetics, 6(7): 1 069-1 077.
DOI:10.1093/hmg/6.7.1069 |
Jiang T, Hou C C, She Z Y, Yang W X. 2013. The SOX gene family:function and regulation in testis determination and male fertility maintenance. Molecular Biology Reports, 40(3): 2 187-2 194.
DOI:10.1007/s11033-012-2279-3 |
Kamachi Y, Uchikawa M, Kondoh H. 2000. Pairing SOX off:with partners in the regulation of embryonic development. Trends in Genetics, 16(4): 182-187.
DOI:10.1016/S0168-9525(99)01955-1 |
Kanai Y, Kanai-Azuma M, Noce T, Saido T C, Shiroishi T, Hayashi Y, Yazaki K. 1996. Identification of two Sox17 messenger RNA isoforms, with and without the high mobility group box region, and their differential expression in mouse spermatogenesis. The Journal of Cell Biology, 133(3): 667-681.
DOI:10.1083/jcb.133.3.667 |
Kanda H, Kojima M, Miyamoto N, Ito M, Takamatsu N, Yamashita S, Shiba T. 1998. Rainbow trout Sox24, a novel member of the Sox family, is a transcriptional regulator during oogenesis. Gene, 211(2): 251-257.
DOI:10.1016/S0378-1119(98)00100-0 |
Kersanach R, Brinkmann H, Liaud M F, Zhang D X, Martin W, Cerff R. 1994. Five identical intron positions in ancient duplicated genes of eubacterial origin. Nature, 367(6461): 387-389.
DOI:10.1038/367387a0 |
Kikuchi Y, Agathon A, Alexander J, Thisse C, Waldron S, Yelon D, Thisse B, Stainier D Y. 2001. casanova encodes a novel Sox-related protein necessary and sufficient for early endoderm formation in zebrafish. Genes & Development, 15(12): 1 493-1 505.
|
Koopman P, Schepers G, Brenner S, Venkatesh B. 2004. Origin and diversity of the SOX transcription factor gene family:genome-wide analysis in Fugu rubripes. Gene, 328: 177-186.
DOI:10.1016/j.gene.2003.12.008 |
Kumar S, Stecher G, Tamura K. 2016. MEGA7:molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7): 1 870-1 874.
DOI:10.1093/molbev/msw054 |
Liu Q Y, Lu H J, Zhang L H, Xie J, Shen W Y, Zhang W M. 2012. Homologues of sox8 and sox10 in the orangespotted grouper Epinephelus coioides:sequences, expression patterns, and their effects on cyp19a1a promoter activities in vitro. Comparative Biochemistry and Physiology Part B:Biochemistry and Molecular Biology, 163(1): 86-95.
DOI:10.1016/j.cbpb.2012.05.004 |
Luckenbach J A, Early L W, Rowe A H, Borski R J, Daniels H V, Godwin J. 2005. Aromatase cytochrome P450:cloning, intron variation, and ontogeny of gene expression in southern flounder (Paralichthys lethostigma). Journal of Experimental Zoology Part A:Comparative Experimental Biology, 303(8): 643-656.
|
Magie C R, Pang K, Martindale M Q. 2005. Genomic inventory and expression of Sox and Fox genes in the cnidarian Nematostella vectensis. Development Genes and Evolution, 215(12): 618-630.
DOI:10.1007/s00427-005-0022-y |
Matsui T, Kanai-Azuma M, Hara K, Matoba S, Hiramatsu R, Kawakami H, Kurohmaru M, Koopman P, Kanai Y. 2006. Redundant roles of Sox17 and Sox18 in postnatal angiogenesis in mice. Journal of Cell Science, 119(17): 3 513-3 526.
DOI:10.1242/jcs.03081 |
Nagai K. 2001. Molecular evolution of Sry and Sox gene. Gene, 270(1-2): 161-169.
DOI:10.1016/S0378-1119(01)00479-6 |
Nakamura S, Watakabe I, Nishimura T, Toyoda A, Taniguchi Y, Tanaka M. 2012. Analysis of medaka sox9 orthologue reveals a conserved role in germ cell maintenance. PLoS One, 7(1): e29982.
DOI:10.1371/journal.pone.0029982 |
Ng L J, Wheatley S, Muscat G E O, Conway-Campbell J, Bowles J, Wright E, Bell D M, Tam P P L, Cheah K S E, Koopman P. 1997. SOX9 binds DNA, activates transcription, and coexpresses with type Ⅱ collagen during chondrogenesis in the mouse. Developmental Biology, 183(1): 108-121.
DOI:10.1006/dbio.1996.8487 |
Ohe K, Tamai K T, Parvinen M, Sassone-Corsi P. 2009. DAX-1 and SOX6 molecular interplay results in an antagonistic effect in pre-mRNA splicing. Developmental Dynamics, 238(6): 1 595-1 604.
DOI:10.1002/dvdy.v238:6 |
Pennisi D, Gardner J, Chambers D, Hosking B, Peters J, Muscat G, Abbott C, Koopman P. 2000. Mutations in Sox18 underlie cardiovascular and hair follicle defects in ragged mice. Nature Genetics, 24(4): 434-437.
DOI:10.1038/74301 |
Popovic J, Stanisavljevic D, Schwirtlich M, Klajn A, Marjanovic J, Stevanovic M. 2014. Expression analysis of SOX14 during retinoic acid induced neural differentiation of embryonal carcinoma cells and assessment of the effect of its ectopic expression on SOXB members in HeLa cells. PLoS One, 9(3): e91852.
DOI:10.1371/journal.pone.0091852 |
Roose J, Korver W, De Boer R, Kuipers J, Hurenkamp J, Clevers H. 1999. The Sox-13 gene:structure, promoter characterization, and chromosomal localization. Genomics, 57(2): 301-305.
DOI:10.1006/geno.1999.5779 |
Saitou N, Nei M. 1987. The neighbor-joining method:a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4(4): 406-425.
|
Sánchez-Soriano N, Russell S. 2000. Regulatory mutations of the Drosophila Sox gene Dichaete reveal new functions in embryonic brain and hindgut development. Developmental Biology, 220(2): 307-321.
DOI:10.1006/dbio.2000.9648 |
Sarraj M A, Wilmore H P, McClive P J, Sinclair A H. 2003. Sox15 is up regulated in the embryonic mouse testis. Gene Expression Patterns, 3(4): 413-417.
DOI:10.1016/S1567-133X(03)00085-1 |
Schepers G E, Teasdale R D, Koopman P. 2002. Twenty Pairs of Sox:extent, homology, and nomenclature of the mouse and human Sox transcription factor gene families. Developmental Cell, 3(2): 167-170.
DOI:10.1016/S1534-5807(02)00223-X |
Shin C H, Chung W S, Hong S K, Ober E A, Verkade H, Field H A, Huisken J, Stainier D Y R. 2008. Multiple roles for Med12 in vertebrate endoderm development. Developmental Biology, 317(2): 467-479.
DOI:10.1016/j.ydbio.2008.02.031 |
Sinclair A H, Berta P, Palmer M S, Hawkins J R, Griffiths B L, Smith M J, Foster J W, Frischauf A M, Lovell-Badge R, Goodfellow P N. 1990. A gene from the human sexdetermining region encodes a protein with homology to a conserved DNA-binding motif. Nature, 346(6281): 240-244.
DOI:10.1038/346240a0 |
Tanimura N, Saito M, Ebisuya M, Nishida E, Ishikawa F. 2013. Stemness-related factor Sall4 interacts with transcription factors Oct-3/4 and Sox2 and occupies Oct-Sox elements in mouse embryonic stem cells. Journal of Biological Chemistry, 288(7): 5 027-5 038.
DOI:10.1074/jbc.M112.411173 |
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley D R, Pimentel H, Salzberg S L, Rinn J L, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3): 562-578.
DOI:10.1038/nprot.2012.016 |
Tsagaratou A, Äijö T, Lio C W J, Yue X, Huang Y, Jacobsen S E, Lähdesmäki H, Rao A. 2014. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proceedings of the National Academy of Sciences of the United States of America, 111(32): E3306-E3315.
DOI:10.1073/pnas.1412327111 |
Vidal V P I, Chaboissier M C, de Rooij D G, Schedl A. 2001. Sox9 induces testis development in XX transgenic mice. Nature Genetics, 28(3): 216-217.
DOI:10.1038/90046 |
Vriz S, Joly C, Boulekbache H, Condamine H. 1996. Zygotic expression of the zebrafish Sox-19, an HMG box-containing gene, suggests an involvement in central nervous system development. Molecular Brain Research, 40(2): 221-228.
DOI:10.1016/0169-328X(96)00052-6 |
Wang R, Cheng H H, Guo Y Q, Zhou R J. 2002. Phylogenic analysis of the Sox gene family of vertebrate. Acta Genetica Sinica, 29(11): 990-994.
(in Chinese with English abstract) |
Wang Y, Ristevski S, Harley V R. 2006. SOX13 exhibits a distinct spatial and temporal expression pattern during chondrogenesis, neurogenesis, and limb development. Journal of Histochemistry & Cytochemistry, 54(12): 1 327-1 333.
|
Wegner M. 2011. SOX after SOX:SOXession regulates neurogenesis. Genes & Development, 25(23): 2 423-2 428.
|
Wei L, Yang C, Tao W J, Wang D S. 2016. Genome-wide identification and transcriptome-based expression profiling of the Sox gene family in the Nile Tilapia(Oreochromis niloticus). International Journal of Molecular Sciences, 17(3): 270.
DOI:10.3390/ijms17030270 |
Weitschek E, Fiscon G, Fustaino V, Felici G, Bertolazzi P. 2015. Clustering and classification techniques for gene expression profile pattern analysis. In: Elloumi M, ↱ Iliopoulos C, ↱ Wang J T L, ↱ Zomaya A Y eds. Pattern Recognition in Computational Molecular Biology: Techniques and Approaches. Wiley, Hoboken, New Jersey. p.347.
|
Wilson M J, Dearden P K. 2008. Evolution of the insect Sox genes. BMC Evolutionary Biology, 8(1): 120.
DOI:10.1186/1471-2148-8-120 |
Wunderle V M, Critcher R, Ashworth A, Goodfellow P N. 1996. Cloning and characterization of SOX5, a new member of the human SOX gene family. Genomics, 36(2): 354-358.
DOI:10.1006/geno.1996.0474 |
Zhang C, Basta T, Klymkowsky M W. 2005. SOX7 and SOX18 are essential for cardiogenesis in Xenopus. Developmental Dynamics, 234(4): 878-891.
DOI:10.1002/dvdy.v234:4 |
Zhang J, Hu Y H, Sun B G, Xiao Z Z, Sun L. 2013. Selection of normalization factors for quantitative real time RTPCR studies in Japanese flounder (Paralichthys olivaceus) and turbot (Scophthalmus maximus) under conditions of viral infection. Veterinary Immunology and Immunopathology, 152(3-4): 303-316.
DOI:10.1016/j.vetimm.2012.12.018 |
Zhang L H, Lin D, Zhang Y, Ma G Z, Zhang W M. 2008. A homologue of Sox11 predominantly expressed in the ovary of the orange-spotted grouper Epinephelus coioides. Comparative Biochemistry and Physiology Part B:Biochemistry and Molecular Biology, 149(2): 345-353.
DOI:10.1016/j.cbpb.2007.10.006 |
Zhang W, Liu Y Z, Yu H Y, Du X X, Zhang Q Q, Wang X B, He Y. 2016. Transcriptome analysis of the gonads of olive flounder (Paralichthys olivaceus). Fish Physiology and Biochemistry, 42(6): 1 581-1 594.
DOI:10.1007/s10695-016-0242-2 |
Zuckerkandl E, Pauling L. 1965. Evolutionary divergence and convergence in proteins. In: Evolving Genes and Proteins.Academic Press, New York. p.97-166.
|