 2019, Vol. 37
2019, Vol. 37Institute of Oceanology, Chinese Academy of Sciences
Article Information
- LI Xiaohong, YOU Cai, QU Liang, ZHOU Bin, TANG Xuexi, XIAO Hui
- Bacterial communities fluctuate in abundance and diversity under simulated oil-contaminated seawater conditions
- Journal of Oceanology and Limnology, 37(2): 615-627
- http://dx.doi.org/10.1007/s00343-019-8039-7
Article History
- Received Mar. 3, 2018
- accepted in principle May. 11, 2018
- accepted for publication May. 30, 2018




2 Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266071, China;
3 Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China;
4 CNOOC Limited, Tianjin Branch, Tianjin 300459, China
Given the frequency of oil spills worldwide, petroleum pollution has become a serious problem in many marine ecosystems. Thus, effective treatments in response to oil pollution has become a primary concern. Typically, mechanical methods, bioremediation, and surfactant chemical methods are used to remove hydrocarbons from contaminated marine sites. Among them, biodegradation through bioremediation processes is the most promising technology since it is more economical and environmentally friendly than other methods (Varjani and Srivastava, 2015). Bacteria are the primary degraders in the natural biodegradation process of hydrocarbons, and as such have become the focus of research on petroleum remediation and treatment (Meckenstock et al., 2016).
Over the past decade, most studies have been conducted on the types of oil-degrading bacteria and their oil degrading efficiency. Many studies focus on which bacterial species were most efficient in oil degradation and under which specific conditions (Yakimov et al., 2007; Mishamandani et al., 2016; Zhou et al., 2016). These studies also showed that oildegrading bacteria typically bloom and become dominant members of the prevailing bacterial communities thus amplifying the bioremediation process (Yakimov et al., 2007; Mishamandani et al., 2016; Zhou et al., 2016). To date, more than 79 genera of oil-degrading bacteria have been identified, and the number of species is still growing (Prince et al., 2010). The efficiency of oil-degrading bacteria can reach approximately 90%, however, it may be affected by many factors including salinity and nutrients (Bai et al., 2007).
Studies have also shown the impact of oil pollution on changes in abundance and structure of bacterial communities (Brakstad and Lødeng, 2005; Cappello et al., 2007). Changes in heterotrophic cultured bacteria, hydrocarbon-degrading bacteria, and total bacterial in oil-contaminated ecosystems have been primarily monitored by culturing techniques or Most Probable Number (MPN) analysis (Brakstad and Lødeng, 2005; Cappello et al., 2007). Previous studies have shown that the growth of bacteria, especially oildegrading bacteria, is stimulated by oil pollution in the initial stages, followed by a variety of trends that range from an overall decline to a series of fluctuations (Brakstad and Lødeng, 2005; Cappello et al., 2007). This was largely due to differences in experimental conditions among the various studies. Likewise, analysis of bacterial communities present in oilcontaminated ecosystems originally relied upon using culture-dependent methods. One downside was that the cultivation approaches used in these studies could only cultivate less than 1% of bacterial species (Amann et al., 1995). However, knowledge of bacterial diversity was greatly improved by advanced molecular technologies (Scopa et al., 2006; Wu et al., 2017). Previous studies indicated that the diversity of bacterial communities is greatly affected by oil pollution, but that the changing trend over time clearly differed. For example, some studies indicated that the diversity of bacterial communities decreased during the first 1–3 weeks (Brakstad and Lødeng, 2005; Zrafi-Nouira et al., 2009), whereas Cappello et al. (2007) reported that the bacterial communities responded with increased diversity during the first 15 d. These discrepancies indicate that there are several factors to be considered in studying bacterial degradation in oil pollution. These factors include type of oil involved, location of the oil exposure, factors in the exposed ecosystem, and the addition of nutrients such as nitrogen, phosphorus, and iron (Viggor et al., 2013; Acosta-González et al., 2015). In most of these studies, reported changes in bacterial community structure after oil pollution designated the bacteria as decomposers. However, the role of bacteria as "producers" in these systems was rarely noticed. Bacterial productivity plays an important role in the energy transfer of marine ecosystems and needs to be considered. The study of bacterial productivity is crucial to understanding material circulation after oil pollution to ascertain the impact of oil pollution on bacterial productivity.
In recent years, in situ tests and laboratory experiments designed to simulate petroleum spill events have been used to study changes in bacterial communities after oil pollution. Due to the frequent occurrence of oil pollution accidents worldwide, in situ investigations have become a common test. Although in situ investigation is preferred, an opensea oil slick is easily dissipated, thus containing the slick for this type of study is not feasible (Brussaard et al., 2010). In addition, this method is susceptible to the effects caused by advection, diffusion, and mixing under natural ocean conditions and is also susceptible to human factors when sampling from inside the experimental spill (Chronopoulou et al., 2015). However, controlled laboratory studies such as microcosms and mesocosms may reduce the influence of uncontrollable environmental variables (Wu et al., 2017), while also allowing for the proper collection and disposal of contaminants at the end of the experiment (Prince, 2015). In terms of microcosms and mesocosms, a microcosm is simple and convenient to operate, but the experimental volume of a microcosm is relatively small, and as such cannot effectively simulate in situ research. In contrast, compared with small-scale laboratory microcosms, mesocosms, which are typically within the range of 1–10 m3 seawater, are closer to real environmental situations and may be optimal in obtaining representative bacterial data isolated from larger volumes (Lebaron et al., 2001).
In this study, a mesocosm experiment was selected to analyze changes in bacterial communities from three different perspectives: quantity, structure, and productivity. Changes in bacterial communities during the biodegradation processes may provide a scientific basis for bioremediation of petroleum pollution.
2 MATERIAL AND METHOD 2.1 MaterialFive liters of Boxi crude petroleum was used in this study as the test oil. The test pool was a marine oil spill weathering process simulation pool which was 5-m long, 3-m wide, 0.4-m high, with a water depth of 0.2 m. Three hundred liters of seawater from the surface of the Shilaoren Sea in Qingdao was collected and used in the test pool.
2.2 Experimental treatmentThe temperature and illumination were kept at 25℃ and 1 000 lux, respectively. Three liters of seawater was collected from the test pool at 0, 7, 14, 21, and 28 d. Fifty milliliters samples were fixed with sterile formaldehyde at a final concentration of 2% (v/v) for cell counts. All samples were stored at 4℃ in the dark, transported to the laboratory for processing and immediately analyzed for bacterial abundance. Samples for bacterial community analysis were filtered through a 0.2-μm filter membrane and then stored at -80℃ until DNA extraction.
2.3 Analysis of bacterial abundance and productivityHeterotrophic plate count (HPC) was used to count the cultured bacteria from collected samples. Colonyforming bacteria were isolated by a marine agar 2216 medium (Xu et al., 1999). Colonies were counted after 7 d of incubation at 28℃. The abundance of total bacteria was determined by using the standard methods of Acridine Orange Direct Counts (AODC) (Xu et al., 1999). The bacterial biomass and volume were also calculated in this study according to following formula.
V=π/4×(L–W/3)×W2,
where V: bacterial volume (μm3); L: bacterial length (μm); W: bacterial width (μm).
BB=8.99×10-8×Vm0.59×BN
where BB: bacterial biomass (μg/L); Vm: bacterial volume (μm3/cell); BN: the number of bacteria (cell/L).
In addition, based on the bacterial biomass and bacterial volume, bacterial productivity was also calculated (Fuhrman and Azam, 1980; Porter and Feig, 1980).
The abundance of oil-degrading bacteria was determined by the Most Probable Number (MPN) method (Wrenn and Venosa, 1996). The medium consisted of (per liter of distilled water): NH4NO3, 1.0 g; K2HPO4, 1.0 g; MgS04·7H2O, 0.1 g; trace amount of FePO4; NaCl, 15 g; No. 20 diesel 1% (W/V); pH 7.2.
2.4 Analysis of change in bacterial community structure at day 0 and day 28 by amplified ribosomal DNA restriction analysis (ARDRA)Samples were collected from the test pool on day 0 and day 28 of the study and subsequently labeled A and B, respectively. Approximately 1 L of sample was filtered through a 0.2-μm pore-size filter membrane. Bacteria were collected on filter membrane and the total genomic DNA was extracted as previously described (Osborn et al., 2000).
Bacterial universal primers were used to amplify the 16S rRNA, namely 5′-AGAGTTTGATCCTGGCTCAG-3′ (27F) and 5′-GGTTACCTTGTTACGACTT-3′ (1492R). DNA amplification was conducted in 50 μL PCR mixture containing 500 μmol/L dNTP, 2 μL of each primer, 20 mmol/L Tris-HCl pH 8.3, 100 mmol/L KCl, 3 mmol/L MgCl2, 0.1 U of Taq polymerase and 2 μL of template DNA. Negative control reactions were executed without DNA. Touchdown PCR was performed to improve specificity of the amplifications. After initial denaturation for 5 min at 95℃, a touch-down PCR was conducted with 20 cycles consisting of 1 min denaturation at 95℃, 1 min annealing at 65℃ and 1 min elongation at 72℃. In the next 20 cycles the temperature of annealing was lowered by approximately 0.5 degrees, followed by 10 cycles of 1 min at 95℃, 1 min at 55℃ and 1 min at 72℃. A final extension step was done for 10 min at 72℃.
The PCR products were subjected to 1% agarose gel electrophoresis. The target DNA bands were extracted with a DNA gel extraction kit (TIANGEN, Beijing) and ligated to the pGM-T Vectors. Then, the ligation mixture was transformed into competent E. coli strain TOP10. Positive clones were screened using ampicillin and by PCR. Primers T7 (5′-TAATACGACTCACTATAGGG-3′) and SP6 (5′-GATTTAGGTGACACTATAG-3′) were used to filtrate positive clones. Five microliters of PCR products were subjected to 1% agarose gel electrophoresis. The PCR products of positive clones were used as template DNA, and 27F and 1492R were used as primers for the second amplification. Finally, PCR products were subjected to restriction enzyme analysis.
The amplified DNA was digested with RsaI and MspI to generate restriction profiles. Restriction analysis of amplified DNA was done with 10 μL of PCR mix, which contained 0.25 μL of the restriction enzyme (RsaI and MspI), 1 μL of 10× NEBuffer (New England Biolabs) and 5 μL of PCR product. Incubations were done for 1 h at 37℃ to ensure complete digestions. Digests were separated on 3% (wt/vol) agarose Tris-Boric acid-EDTA (TBE) gels and analyzed.
2.5 Data analysisRestriction patterns of individual isolates and clones were handled by the software of DNASTAR. Lasergene.v7.1 and SeqMed program. In addition, CHIMERA-CHECK was also used to eliminate chimera sequences. The final sequences were compared to known sequences in GenBank. The most similar sequences of each construct were selected for DNA sequence comparison by using the Clustal X (1.8) software. Shannon-Weiner, Evenness, and Simpson indices and the Margalef index were calculated as described (Elshahed et al., 2003). Diversity coverage was also calculated as described by Good (1953).
3 RESULT 3.1 Changes in bacterial abundance and productivityChanges in abundance of cultured heterotrophic bacteria and total bacteria showed a similar trend during the 28 d of the experiment. The trend involved a decrease in the first 7-d, then a slight increase from the 7th to the 14th day, followed by relatively low steady-state levels afterwards. The number of cultured heterotrophic bacteria and total bacteria ranged from 2.9×105cfu/mLto 7.5×105cfu/mL and 5.4×1010cell/mL to 1.1×1011 cell/mL, respectively. Changes in the abundance of bacterial biomass (3.9×102 μg/L to 5.3×102 μg/L) showed a similar trend with that of cultured heterotrophic bacteria and total bacteria. However, changes in the trend of bacterial volume and bacterial productivity (calculated according to bacterial biomass and bacterial volume) were inconsistent. The levels of bacterial volume and bacterial productivity increased with time, and peaked on the 7th day at 0.1×10-1 μm3 and 40.92 μg C/(L·h) from 0.7×10-2 μm3 and 22.19 μgC/(L·h), respectively (Fig. 1). The partial fluorescence micrograph of bacteria is shown in Suppl. Fig. 1.

|
| Fig.1 Changes in the abundance of cultured heterotrophic bacteria and total bacteria (a), bacterial biomass and bacterial productivity (b), bacterial volume (c) |
Besides, the abundance of oil-degrading bacteria increased with time and reached a peak of 9.5×102 cell/mL on the 21st day with a subsequent decline later in the period. In addition, a similar trend was observed in the ratio of oil-degrading bacteria to the total number of bacteria (Fig. 2).

|
| Fig.2 Changes in the abundance of oil-degrading bacteria (a) and the ratio of oil-degrading bacteria to the total number of bacteria (b) |
The 16S rRNA PCR reaction products of samples A and B were 1 500 bp. The 16 s rDNA PCR amplification results of samples A and B are shown in Suppl. Fig. 2. A1-A4 were the 16 s rRNA PCR amplifications of sample A, and B1-B4 were the 16 s rDNA PCR amplifications of sample B.
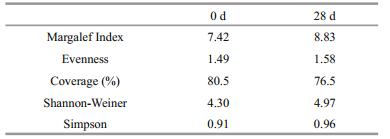
3.2.2 The construction of 16S rRNA clone librariesTwo hundred clones from each sample were randomly selected for positive clones. Libraries A and B yielded 167 and 183 positive clones, respectively. The coverage of the two libraries were 80.5% and 76.5%, which may reflect the bacterial diversity of the samples (Table 1). For sample A, the Margalef index was 7.42, there was an Evenness of 1.49, and the Shannon-Weiner and Simpson indices were 4.30 and 0.91, respectively. For sample B, the Margalef index was 8.83, there was an Evenness of 1.58, and the Shannon-Weiner and Simpson indices were 4.97 and 0.96, respectively (Table 1). The electrophoretic profile of partial 16S rRNA PCR amplification of screened positive clones is shown in Suppl. Fig. 3. The size of fragments was approximately 1 600 bp.

|
| Fig.3 Partial ARDRA patterns of sample A (a) and sample B (b) obtained after restriction of the amplified 16S rRNA gene with two different enzymes (RsaI and MspI) |
As shown in Fig. 3, 157 and 183 positive clones of samples A and B were digested by two restriction endonucleases: RsaI and MspI. There were 39 and 60 different restriction patterns obtained from samples A and B, respectively.
3.3 Bacterial community structures in sample AAt phylum level, three prokaryotic phyla were found in sample A: Proteobacteria (80.84%), Bacteroidetes (14.37%) and Actinobacteria (4.79%). Proteobacteria was the dominant phylum, followed by Bacteroidetes. At the class level, six classes of prokaryotes were found overall (Fig. 4a). Of the entire bacterial community, Epsilonproteobacteria (51.10%) was the most abundant class, with Alphaproteobacteria (17.96%) and Betaproteobacteria (7.19%) being the second and third most abundant. Sixteen genera were identified (Fig. 5a). Arcobacter (51.50%) was the most abundant, followed by Lentibacter (8.98%) and Lutibacter (7.78%). Other genera were well represented, such as Aquimarina (5.99%) and Aquiluna (4.79%). Based on these data, a representative of each genus was selected to perform a phylogenetic analysis for 16S rRNA gene sequence analysis (Fig. 6a).

|
| Fig.4 Relative abundance of bacterial 16S rRNA gene sequences presented at the class level a. 0 d; b. 28 d. |

|
| Fig.5 Relative abundance of bacterial 16S rRNA gene sequences presented at the genus level a. 0 d; b. 28 d. |

|
| Fig.6 Neighbor joining phylogenetic trees based on 16S rRNA sequences (a representative of each genus) of the bacteria a. sample A and related GenBank species; b. sample B and related GenBank species. Numerical values indicate bootstrap percentile from 1 000 replicates. |
At phylum level, three prokaryotic phyla were found in sample B: Proteobacteria (97.81%), Bacteroidetes (1.64%) and Actinobacteria (0.55%). At the class level, eight prokaryotes classes were found (Fig. 4b). Of the entire bacterial community, Alphaproteobacteria (40.98%) was the most abundant class, with Gammaproteobacteria (38.80%) and Betaproteobacteria (13.66%) being the second and the third most dominant classes, respectively. There were 32 genera identified (Fig. 5b). Legionella (16.94%) was the most abundant genus, followed by Methylotenera (11.48%) and then Cycloclasticus (10.38%). Other genera were well represented, such as Oceanibaculum (9.84%). They all appeared regularly in oil-contaminated environments and were regarded as oil-degrading bacteria. In addition, some other bacteria related to petroleum-degradation also appeared, such as Thalassospira (2.73%), and Alteromonas spp. (1.64%). These were not found in sample A. Furthermore, based on these data, a representative of each genus was also selected to perform a phylogenetic analysis for 16S rRNA gene sequence analysis (Fig. 6b).
In this study some species, which were regarded as oil-degrading bacteria in previous studies, emerged 28 d after oil pollution. These organisms included Cycloclasticus (Yakimov et al., 2007), Thalassospira (Zhou et al., 2016), Porticoccus (Gutierrez et al., 2012), Arenibacter (Mishamandani et al., 2016), Sulfurimonas (Sun et al., 2017), Pusillimonas (Lladó et al., 2013) and Azoarcus (Ruan et al., 2016). Among them, Cycloclasticus, Thalassospira, and Porticoccus are common oil-degrading bacteria.
4 DISCUSSION 4.1 Analysis of changes in abundance and bacterial productivityRecent studies have shown that bacterial communities in oil-contaminated ecosystems are greatly affected by oil pollution. In this study, there was a rapid decline in the abundance of bacteria from day zero to day seven. However, previous studies have shown that the abundance of bacteria increases in the initial stages of petroleum pollution (Hassanshahian et al., 2014; Wu et al., 2017). One significant difference is that in previous studies nutrients were added to oil-contaminated seawater. The addition of nutrients provided the necessary conditions for significant bacterial growth, leading to an increase in the abundance of bacteria (Röling et al. 2002). However, to accurately simulate the natural environment, we performed the experiment without adding additional nutrients to the seawater. The decreased bacterial abundance may also be caused by the toxicity of the oil used, as different types of oil may have different effects (Zanaroli et al., 2010). Moreover, the change in trend of bacterial productivity increased with time in the beginning stages. Piehler et al. (2002) also reported that bacterial productivity was elevated to some extent after adding diesel fuel. Chronic petroleum pollution has been found to have different effects on bacterial productivity, ranging from stimulation to inhibition (Montagna et al., 1987; Long et al., 1995). It is well known that bacterial productivity is closely linked to bacterial biomass and bacterial volume. As bacterial volume and productivity have a consistent changing trend, we speculate that bacterial volume has a greater impact on productivity than bacterial biomass. The increase in bacterial volume was caused by the absorption and utilization of oil components.
Furthermore, the abundance of oil-degrading bacteria increased apparently in the initial stage and was followed with a subsequent decline later in the period. The proportion of oil-degrading bacteria to the number of cultured heterotrophic bacteria increased from 0.1% to 30.75% (Fig. 2), which is consistent with previous reports (Atlas, 1981, 1991; de la Cueva et al., 2016). Oil-degrading bacteria accounted for less than 0.1% of the total population in unpolluted ecosystems. However, oil-degrading bacteria were often seen to increase from 1%–10% of the microbial communities in an environment contaminated with oil pollution, even increasing to 100% of the total viable bacteria (Atlas, 1981, 1991; Jean et al., 2008). The proportion of oil-degrading bacteria rose in varying degrees depending on factors such as differences in bacterial types, variations in the amount of oil, and changing environmental conditions. The percentage of oil-degrading bacteria may reflect the degree or extent of exposure to hydrocarbon contaminants (Atlas, 1981).
4.2 Analysis of changes in the bacterial community structureNumerous research studies have indicated that bacterial diversity is affected by oil contamination (Liao et al., 2015; de la Cueva et al., 2016). Most studies observed a decrease in bacterial biodiversity after oil pollution due to the toxic effects of petroleum on bacterial communities (Zrafi-Nouira et al., 2009; de la Cueva et al., 2016). Conversely, some studies found that the bacterial biodiversity increased (dos Santos et al., 2011; Liao et al., 2015), which was like our study. First, the types of oil affected the diversity of bacterial communities as a single species that can only biodegrade a limited range of petroleum hydrocarbons (Zanaroli et al., 2010). In this study, the experimental oil was crude oil whose composition is complex and leads to a wider range of oildegrading bacteria resulting in higher bacterial diversity. Liao et al. (2015) also found that the bacterial biodiversity was significantly increased after Daqing oil and Huabei oil contamination. However, either diesel or gasoline is typically used, representing a relatively simple composition. Second, the concentration of oil had a great influence on the diversity of bacterial communities. Dos Santos et al. (2011) reported that both 2% and 5% oil could increase the bacterial diversity, but that the change caused by 5% oil was reduced. This observation was probably due to the toxicity of the higher oil content. Third, the size of the experiment also had a certain impact on the experimental results. Compared with microcosm, mesocosm is more natural and semi-realistic since it has an indigenous combination of abiotic conditions and organisms, and has an increase of the degree of niches in the system (dos Santos et al., 2011; Liao et al., 2015). Finally, season and nutrient conditions of oil contamination may also lead to differences in changes in bacterial biodiversity (Liao et al., 2015).
Compared to previous findings, the bacterial community composition of sample A was different at class and genus levels. At the class level, Epsilonproteobacteria was the most dominant bacteria accounting for 51.10% of the total, which differed from that reported in previous studies (Li et al., 2006; Xiao et al., 2009; Jiang et al., 2014). Li et al. (2006) and Xiao et al. (2009) reported that Gammaproteobacteria was the dominant bacteria in the coastal waters of Qingdao, but Alphaproteobacteria, Betaproteobacteria and Deltaproteobacteria were reported as the dominant bacteria in the seawater in Laizhou Bay (Jiang et al., 2014). At the genus level, it is more difficult to unify the results of surveys of bacterial community composition in different sea areas. Li et al. (2006) showed that Vibrio was the dominant genus in the clean sea environment of Qingdao. However, Vibrio and Acinetobacter were the dominant genera in the seawater near the industrial area of Qingdao. Likewise, Vibrio and Pseudoalteromonas were the dominant genera of seawater near breeding intensive areas of Qingdao. The findings of bacterial community composition are difficult to unify due to the difference in sampling time, sampling specific locations, and methodological limitations. In this study, there were many common bacteria in the marine environment, such as Pseudomonas, Vibrio, and Bacillus. However, Arcobacter, Lentibacter, and Lutibacter were the most abundant in sample A. Among them, Arcobacter, which is a relatively poorly known group of bacteria as it is difficult to cultivate, was the dominant genus. But, the primary source of new Arcobacter species seems to be marine ecosystems (Collado and Figueras, 2011). Lentibacter was discovered from the seawater in the Qingdao intertidal zone by Li et al. (2014). The experimental results here were different from the previous study due to chance errors of sampling. The dominant bacteria found in our experiments were unusual bacteria, but these dominant bacteria have previously appeared in the marine ecosystem.
Many factors can affect bacterial community composition, such as contaminants, temperature, and pH. Among them, pollution is one of the important factors (Liao et al., 2015; de la Cueva et al., 2016). The composition of bacterial communities changes significantly after oil pollution. In this study, at the class level, Alphaproteobacteria (40.98%) replaced Epsilonproteobacteria (51.10%) as the most abundant class, and Gammaproteobacteria (38.80%) became the second most dominant class in the whole bacterial community. These results are consistent with previous studies, which also indicated that the abundance of Alphaproteobacteria and Gammaproteobacteria increased and became the dominant classes after oil pollution (Viggor et al., 2013; Acosta-González et al., 2015). For example, Mason et al. (2014) found that Gammaproteobacteria significantly increased its relative abundance within the whole bacterial community after oil pollution. Gammaproteobacteria includes the majority of oil-degrading bacteria (Bælum et al., 2012), has a strong adaptability, and is widely distributed in different ecosystems. In addition, many bacterial species of γ-Proteobacteria are involved in the metabolism of sulfur, ammonia, and methyl along with the degradation of high molecular weight polycyclic aromatic hydrocarbon (PAHs) (Brambilla et al., 2001). At the genus level, the number of genera in sample B were nearly twice as much as that of sample A where Legionella, Methylotenera, and Cycloclasticus were the dominant genera. Legionella, Methylotenera and Cycloclasticus have been reported to be related to the degradation of organic matter (Kalyuzhnaya et al., 2006; Yakimov et al., 2007; Ding et al., 2012). Legionella was also enriched in Luvisol soil which was polluted by phenanthrene (a model compound for PAH) (Ding et al., 2012). Methylotenera was also the dominant genus in oil contaminated soils and is known as an obligate methyl utilizer (Kalyuzhnaya et al., 2006; Jiao et al., 2016). Moreover, Cycloclasticus is already regarded as the obligate marine PAH degrader (Yakimov et al., 2007) and is an important genus found in oil-contaminated marine environments (Dong et al., 2015). Some other oil-degrading bacteria, which were detected in oil-contaminated environments previously, were also found in this study, such as Thalassospira, Arenibacter, and Altererythrobacter (Mishamandani et al., 2016; Zhou et al., 2016; Wu et al., 2017).
4.3 Analysis of the occurrence of oil-degrading bacteriaStudies have shown that a variety of oil-degrading bacteria will proliferate after an oil spill event. Different types of oil-degrading bacteria represented in a spill depend on the types of oil and the location of oil exposure. In this study, some species, which were regarded as oil-degrading bacteria in previous studies, emerged 28 d after oil pollution. These organisms included, Cycloclasticus (Yakimov et al., 2007), Thalassospira (Zhou et al., 2016), Porticoccus (Gutierrez et al., 2012), Arenibacter (Mishamandani et al., 2016), Sulfurimonas (Sun et al., 2017), Pusillimonas (Lladó et al., 2013) and Azoarcus (Ruan et al., 2016). Among them, Cycloclasticus, Thalassospira, and Porticoccus are common oildegrading bacteria. The oil-degrading bacteria Cycloclasticus pugetii was first reported in 1995. Then, a subsequent study revealed that Cycloclasticus pugetii, or its homologous species, was the primary species of oil-degrading bacteria in the two weeks following a cruise ship accident (Maruyama et al., 2003). Cycloclasticus pugetii degrades aromatic hydrocarbons, naphthalene, phenanthrene, anthracene, pyrene and other polycyclic aromatics. Thalassospira spp. were also identified in oilcontaminated seawater (Liu and Liu, 2013). Some relevant reports have confirmed that Thalassospira spp. were capable of degrading PAHs and contributed significantly to the degradation of aliphatic hydrocarbons (Nogi et al., 2014; Zhou et al., 2016). In addition, Porticoccus, which can utilize hydrocarbons as a unique source of carbon and energy rather than other naturally occurring organic substrates (Gutierrez et al., 2012), was also found in our study. Given the improvement of research methods, and the expansion of the research scope, the types of oil degradation bacteria involved in bioremediation will be better understood.
4.4 Analysis of the use of different experimental scaleThe bacterial community composition changed greatly after oil pollution no matter what kind of experimental scale was used. However, a uniform result was not feasible using different experimental methods. In our study, after 28 d of oil pollution, the diversity of bacterial community composition increased significantly. However, Zrafi-Nouira et al. (2009) observed a clear decrease in bacterial diversity after 28 d of pollution. This may be due in part to the difference in experimental scale. For example, in the present study the mesocosm experiment was used, however Zrafi-Nouira et al. (2009) chose a smallscale laboratory experiment. In addition, the experimental results comparing in situ investigations and mesocosm experiments are also inconsistent. This may be due to the lack of control under in situ conditions. These conditions include: a greater depth of sampled water, dilution factors and the flow of seawater from uncontaminated areas. For example, Chronopoulou et al. (2015) performed a comparative study of bacterial community changes using in situ experiments and mesocosm experiments. Different experimental results were obtained from each method. There were no oil-induced changes in the bacterial community after the experimental spill at sea, whereas the bacterial community of the mesocosm changed significantly. Even with the same experimental method (mesocosm) as used here, the experimental results can still be inconsistent (Nishimura et al., 2006). This may be due to other factors affecting the experimental results, such as different periods of investigation and different types of experimental oil used in the study (Viggor et al., 2013). Even when homogenizing experimental conditions, the experimental results from different scales are inconsistent. This indicates that research in this area needs to be further studied. Important questions regarding specific applications of the different scales, the connection among results, and correlations among outcomes need to be addressed.
5 CONCLUSIONChanges in bacterial abundance and bacterial species in oil-contaminated seawater during the initial stages of pollution under simulated natural conditions were analyzed in this study. The results showed that changes in the abundance of cultured heterotrophic bacteria and total bacteria had a similar trend; there was an initial increase, then a decrease, followed by a relatively low steady-state level thereafter. The changes in the abundance of bacterial biomass also showed a similar trend with those of cultured heterotrophic bacteria and total bacteria. However, the bacterial productivity calculated based on bacterial biomass and bacterial volume showed a different trend, like that of bacterial volume. In addition, the bacterial community also changed greatly after oil pollution. The first and second dominant classes changed from Epsilonproteobacteria and Alphaproteobacteria to Alphaproteobacteria and Gammaproteobacteria. The number of genera increased significantly after oil pollution. Moreover, some bacteria related to petroleum-degrading appeared in the oil-contaminated seawater.
6 DATA AVAILABILITY STATEMENTThe sequences obtained in this study have been deposited in the GenBank database under accession numbers JQ712030–JQ712068 and JQ712069–JQ712128. The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary materialSupplementary material (Supplementary Figs.1–3) is available in the online version of this article at https://doi.org/10.1007/s00343-019-8039-7.
Acosta-González A, Martirani-von Abercron S M, Rosselló-Móra R, Wittich R M, Marqués S. 2015. The effect of oil spills on the bacterial diversity and catabolic function in coastal sediments: a case study on the Prestige oil spill. Environmental Science and Pollution Research, 22(20): 15200-15214.
DOI:10.1007/s11356-015-4458-y |
Amann R I, Ludwig W, Schleifer K H. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiological Reviews, 59(1): 143-169.
|
Atlas R M. 1981. Microbial degradation of petroleum hydrocarbons: an environmental perspective. Microbiological Reviews, 45(1): 180-209.
|
Atlas R M. 1991. Microbial hydrocarbon degradationbioremediation of oil spills. Journal of Chemical Technology and Biotechnology, 52(2): 149-156.
|
Bælum J S, Borglin S, Chakraborty R, Fortney J L, Lamendella R, Mason O U, Auer M, Zemla M, Bill M, Conrad M E, Malfatti S A, Tringe S G, Holman H Y, Hazen T C, Jansson J K. 2012. Deep-sea bacteria enriched by oil and dispersant from the Deepwater Horizon spill. Environmental Microbiology, 14(9): 2405-2416.
DOI:10.1111/emi.2012.14.issue-9 |
Bai J, Cui A L, Lv Y H. 2007. The degradation capability of crude oil degrading strains and the impacting factors. Transactions of Oceanology and Limnology, (3): 41-48.
(in Chinese with English abstract) |
Brakstad O G, Lødeng A G G. 2005. Microbial diversity during biodegradation of crude oil in seawater from the North Sea. Microbial Ecology, 49(1): 94-103.
DOI:10.1007/s00248-003-0225-6 |
Brambilla E, Hippe H, Hagelstein A, Tindall B J, Stackebrandt E. 2001. 16S rDNA diversity of cultured and uncultured prokaryotes of a mat sample from Lake Fryxell, McMurdo Dry Valleys, Antarctica. Extremophiles, 5(1): 23-33.
DOI:10.1007/s007920000169 |
Brussaard C P D, Peperzak L, Witte Y, Huisman J. 2010. An experimental oil spill at sea. In: Timmis K N ed. Handbook of Hydrocarbon and Lipid Microbiology. Springer, Berlin, Heidelberg. p.3 491-3 502.
|
Cappello S, Caruso G, Zampino D, Monticelli L S, Maimone G, Denaro R, Tripodo B, Troussellier M, Yakimov M, Giuliano L. 2007. Microbial community dynamics during assays of harbour oil spill bioremediation: a microscale simulation study. Journal of Applied Microbiology, 102(1): 184-194.
DOI:10.1111/jam.2007.102.issue-1 |
Chronopoulou P, Sanni G O, Silas-Olu D I, Meer J R, Timmis K N, Brussaard C P D, McGenity T J. 2015. Generalist hydrocarbon-degrading bacterial communities in the oilpolluted water column of the North Sea. Microbial Biotechnology, 8(3): 434-447.
DOI:10.1111/mbt2.2015.8.issue-3 |
Collado L, Figueras M J. 2011. Taxonomy, epidemiology, and clinical relevance of the genus Arcobacter. Clinical Microbiology Reviews, 24(1): 174-192.
DOI:10.1128/CMR.00034-10 |
de la Cueva S C, Rodríguez C H, Cruz N O S, Contreras J A R, Miranda J L. 2016. Changes in bacterial populations during bioremediation of soil contaminated with petroleum hydrocarbons. Water, Air, & Soil Pollution, 227: 91.
|
Ding G C, Heuer H, Smalla K. 2012. Dynamics of bacterial communities in two unpolluted soils after spiking with phenanthrene: soil type specific and common responders. Frontiers in Microbiology, 3: 290.
|
Dong C, Bai X, Sheng H, Jiao L, Zhou H, Shao Z. 2015. Distribution of pahs and the pah-degrading bacteria in the deep-sea sediments of the high-latitude arctic ocean. Biogeosciences, 12(7): 2163-2177.
DOI:10.5194/bg-12-2163-2015 |
dos Santos H F, Cury J C, do Carmo F L, dos Santos A L, Tiedje J, van Elsas J D, Rosado A S, Peixoto R S. 2011. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution. PLoS One, 6(3): e16943.
DOI:10.1371/journal.pone.0016943 |
Elshahed M S, Senko J M, Najar F Z, Kenton S M, Roe B A, Dewers T A, Spear J R, Krumholz L R. 2003. Bacterial diversity and sulfur cycling in a mesophilic sulfide-rich spring. Applied and Environmental Microbiology, 69(9): 5609-5621.
DOI:10.1128/AEM.69.9.5609-5621.2003 |
Fuhrman J A, Azam F. 1980. Bacterioplankton secondary production estimates for coastal waters of British Columbia, Antarctica, and California. Applied and Environmental Microbiology, 39(6): 1085-1095.
|
Good I J. 1953. The population frequencies of species and the estimation of population parameters. Biometrika, 40(3-4): 237-264.
DOI:10.1093/biomet/40.3-4.237 |
Gutierrez T, Nichols P D, Whitman W B, Aitken M D. 2012. Porticoccus hydrocarbonoclasticus sp. nov., an aromatic hydrocarbon-degrading bacterium identified in laboratory cultures of marine phytoplankton. Applied and Environmental Microbiology, 78(3): 628-637.
DOI:10.1128/AEM.06398-11 |
Hassanshahian M, Yakimov M M, Denaro R, Genovese M, Cappello S. 2014. Using real-time PCR to assess changes in the crude oil degrading microbial community in contaminated seawater mesocosms. International Biodeterioration & Biodegradation, 93: 241-248.
|
Jean J S, Lee M K, Wang S M, Chattopadhyay P, Maity J P. 2008. Effects of inorganic nutrient levels on the biodegradation of benzene, toluene, and xylene (BTX) by Pseudomonas spp. in a laboratory porous media sand aquifer model. Bioresource Technology, 99(16): 7807-7815.
DOI:10.1016/j.biortech.2008.01.064 |
Jiang H C, Liu A Y, Ren L H, Song X K, Jiang X Y, Liu L J. 2014. PCR-RFLP analysis of bacteria 16S rDNA in Laizhou Bay. Transactions of Oceanology and Limnology, (3): 127-134.
(in Chinese with English abstract) |
Jiao S, Liu Z S, Lin Y B, Yang J, Chen W M, Wei G H. 2016. Bacterial communities in oil contaminated soils: biogeography and co-occurrence patterns. Soil Biology and Biochemistry, 98: 64-73.
DOI:10.1016/j.soilbio.2016.04.005 |
Kalyuzhnaya M G, Bowerman S, Lara J C, Lidstrom M E, Chistoserdova L. 2006. Methylotenera mobilis gen. nov., sp. nov., an obligately methylamine-utilizing bacterium within the family Methylophilaceae. International Journal of Systematic and Evolutionary Microbiology, 56(12): 2819-2823.
DOI:10.1099/ijs.0.64191-0 |
Lebaron P, Servais P, Troussellier M, Courties C, Muyzer G, Bernard L, Schäfer H, Pukall R, Stackebrandt E, Guindulain T, Vives-Rego J. 2001. Microbial community dynamics in Mediterranean nutrient-enriched seawater mesocosms: changes in abundances, activity and composition. FEMS Microbiology Ecology, 34(3): 255-266.
DOI:10.1111/fem.2001.34.issue-3 |
Li P P, Chen X C, Zhang Y R, Zhang X J, Mei G M, Guo Y M. 2014. Purification and structural elucidation of exoploysaccharide from a new marine bacterium lentibacter algarum zxm100t. Chinese Journal of Biotechnology, 30(3): 455-463.
(in Chinese with English abstract) |
Li Y, Zhou H X, Liu J L, Zhang Y F, Chen J X, Zhang X H. 2006. Distribution and identification of marine heterotrophic bacteria in different sea areas close to the shore of Qingdao. Periodical of Ocean University of China, 36(6): 965-970.
(in Chinese with English abstract) |
Liao J Q, Wang J, Jiang D L, Wang M C, Huang Y. 2015. Longterm oil contamination causes similar changes in microbial communities of two distinct soils. Applied Microbiology and Biotechnology, 99(23): 10299-10310.
DOI:10.1007/s00253-015-6880-y |
Liu Z F, Liu J Q. 2013. Evaluating bacterial community structures in oil collected from the sea surface and sediment in the northern Gulf of Mexico after the Deepwater Horizon oil spill. Microbiology Open, 2(3): 492-504.
DOI:10.1002/mbo3.2013.2.issue-3 |
Lladó S, Gràcia E, Solanas A M, Viñas M. 2013. Fungal and bacterial microbial community assessment during bioremediation assays in an aged creosote-polluted soil. Soil Biology and Biochemistry, 67: 114-123.
DOI:10.1016/j.soilbio.2013.08.010 |
Long S C, Aelion C M, Dobbins D C, Pfaender F K. 1995. A comparison of microbial community characteristics among petroleum-contaminated and uncontaminated subsurface soil samples. Microbial Ecology, 30(3): 297-307.
|
Maruyama A, Ishiwata H, Kitamura K, Sunamura M, Fujita T, Matsuo M, Hiqashihara T. 2003. Dynamics of microbial populations and strong selection for Cycloclasticus pugetii following the Nakhodka oil spill. Microbial Ecology, 46(4): 442-453.
DOI:10.1007/s00248-002-3010-z |
Mason O U, Scott N M, Gonzalez A, Robbinspianka A, B?lum J, Kimbrel J, Bouskill N J, Prestat E, Borglin S, Joyner D C, Fortney J L, Jurelevicius D, Stringfellow W T, Alvarez-Cohen L, Hazen T C, Knight R, Gilbert J A, Jansson J K. 2014. Metagenomics reveals sediment microbial community response to Deepwater Horizon oil spill. The ISME Journal, 8(7): 1464-1475.
DOI:10.1038/ismej.2013.254 |
Meckenstock R U, Boll M, Mouttaki H, Koelschbach J S, Tarouco P C, Weyrauch P, Dong X Y, Himmelberg A M. 2016. Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. Journal of Molecular Microbiology and Biotechnology, 26(1-3): 92-118.
DOI:10.1159/000441358 |
Mishamandani S, Gutierrez T, Berry D, Aitken M D. 2016. Response of the bacterial community associated with a cosmopolitan marine diatom to crude oil shows a preference for the biodegradation of aromatic hydrocarbons. Environmental Microbiology, 18(6): 1817-1833.
DOI:10.1111/1462-2920.12988 |
Montagna P A, Bauer J E, Toal J, Hardin D, Spies R B. 1987. Temporal variability and the relationship between benthic meiofaunal and microbial populations of a natural coastal petroleum seep. Journal of Marine Research, 45(3): 761-789.
DOI:10.1357/002224087788326894 |
Nishimura M, Yoshida A, Toyoda K, Yamada M, Nomura H, Wada M, Okamoto K, Shibata A, Takada H, Ohwada K. 2006. Mesocosm experiment on the succession of microbial community in response to oil contamination to coastal seawater. La Mer, 44(2): 59-65.
|
Nogi Y, Yoshizumi M, Miyazaki M. 2014. Thalassospira povalilytica sp. nov., a polyvinyl-alcohol-degrading marine bacterium. International Journal of Systematic and Evolutionary Microbiology, 64(4): 1149-1153.
|
Osborn A M, Moore E R, Timmis K N. 2000. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environmental Microbiology, 2(1): 39-50.
DOI:10.1046/j.1462-2920.2000.00081.x |
Piehler M F, Maloney J S, Paerl H W. 2002. Bacterioplanktonic abundance, productivity and petroleum hydrocarbon biodegradation in marinas and other coastal waters in North Carolina, USA. Marine Environmental Research, 54(2): 157-168.
DOI:10.1016/S0141-1136(02)00101-0 |
Porter K G, Feig Y S. 1980. The use of DAPI for identifying and counting aquatic microflora1. Limnology and Oceanography, 25(5): 943-948.
DOI:10.4319/lo.1980.25.5.0943 |
Prince R C, Gramain A, Mcgenity T J. 2010. Prokaryotic hydrocarbon degraders. In: Timmis K N ed. Handbook of Hydrocarbon and Lipid Microbiology. Springer Berlin Heidelberg.
|
Prince R C. 2015. Introduction: mesocosms and microcosms. In: McGenity T, Timmis K, Nogales B eds. Hydrocarbon and Lipid Microbiology Protocols. Springer Protocols Handbooks. Springer, Berlin, Heidelberg
|
Röling W F M, Milner M G, Jones D M, Lee K, Daniel F, Swannell R J P, Head I M. 2002. Robust hydrocarbon degradation and dynamics of bacterial communities during nutrient-enhanced oil spill bioremediation. Applied and Environmental Microbiology, 68(11): 5537-5548.
DOI:10.1128/AEM.68.11.5537-5548.2002 |
Ruan Y J, Deng Y L, Guo X S, Timmons M B, Lu H F, Han Z Y, Ye Z Y, Shi M M, Zhu S M. 2016. Simultaneous ammonia and nitrate removal in an airlift reactor using poly (butylene succinate) as carbon source and biofilm carrier. Bioresource Technology, 216: 1004-1013.
DOI:10.1016/j.biortech.2016.06.056 |
Scopa A, Salzano G, Scrano L, Bufo S A, Bonomo M G. 2006. Preliminary assessment of microbial community recovery after an accidental oil spill by molecular analysis. Fresenius Environmental Bulletin, 7(1): 675-681.
|
Sun Y J, Lu S D, Zhao X H, Ding A Z, Wang L. 2017. Longterm oil pollution and in situ microbial response of groundwater in Northwest China. Archives of Environmental Contamination and Toxicology, 72(4): 519-529.
DOI:10.1007/s00244-017-0405-x |
Varjani S J, Srivastava V K. 2015. Green technology and sustainable development of environment. Renewable Res earch J ournal, 3(1): 244-249.
|
Viggor S, Juhanson J, Jõesaar M, Mitt M, Truu J, Vedler E, Heinaru A. 2013. Dynamic changes in the structure of microbial communities in Baltic Sea coastal seawater microcosms modified by crude oil, shale oil or diesel fuel. Microbiological Research, 168(7): 415-427.
DOI:10.1016/j.micres.2013.02.006 |
Wrenn B A, Venosa A D. 1996. Selective enumeration of aromatic and aliphatic hydrocarbon degrading bacteria by a most-probable-number procedure. Canadian Journal of Microbiology, 42(3): 252-258.
DOI:10.1139/m96-037 |
Wu M L, Ye X Q, Chen K L, Li W, Yuan J, Jiang X. 2017. Bacterial community shift and hydrocarbon transformation during bioremediation of short-term petroleumcontaminated soil. Environmental Pollution, 223: 657-664.
DOI:10.1016/j.envpol.2017.01.079 |
Xiao H, Zhang Y, Zhang Z, Zhu L, You C, Tang X X. 2009. A preliminary study on the bacterial diversity in surface sediments from the coastal water of Qingdao and Weihai in summer and winter. Periodical of Ocean University of China, 39(4): 641-646.
(in Chinese with English abstract) |
Xu H S, Yang X S, Li Y. 1999. Diagnosis and Control of Bacterial Diseases in Penaeid Shrimp Hatcheries. China Ocean Press, Beijing, China. p.55-101.
(in Chinese)
|
Yakimov M, Timmis K N, Golyshin P N. 2007. Obligate oildegrading marine bacteria. Current Opinion in Biotechnology, 18(3): 257-266.
DOI:10.1016/j.copbio.2007.04.006 |
Zanaroli G, Di Toro S, Todaro D, Varese G C, Bertolotto A, Fava F. 2010. Characterization of two diesel fuel degrading microbial consortia enriched from a non acclimated, complex source of microorganisms. Microbial Cell Factories, 9: 10.
DOI:10.1186/1475-2859-9-10 |
Zhou H Y, Wang H, Huang Y, Fang T T. 2016. Characterization of pyrene degradation by halophilic Thalassospira sp. strain tsl5-1 isolated from the coastal soil of Yellow Sea, China. International Biodeterioration & Biodegradation, 107: 62-69.
|
Zrafi-Nouira I, Guermazi S, Chouari R, Safi N M D, Pelletier E, Bakhrouf A, Saidane-Mosbahi D, Sghir A. 2009. Molecular diversity analysis and bacterial population dynamics of an adapted seawater microbiota during the degradation of Tunisian zarzatine oil. Biodegradation, 20(4): 467-486.
DOI:10.1007/s10532-008-9235-x |