 2019, Vol. 37
2019, Vol. 37Institute of Oceanology, Chinese Academy of Sciences
Article Information
- DING Haiyan, GUO Li, LI Xiaojie, YANG Guanpin
- Transcriptome analysis of kelp Saccharina japonica unveils its weird transcripts and metabolite shift of main components at different sporophyte developmental stages
- Journal of Oceanology and Limnology, 37(2): 640-650
- http://dx.doi.org/10.1007/s00343-019-8019-y
Article History
- Received Feb. 1, 2018
- accepted in principle May. 3, 2018
- accepted for publication Jun. 25, 2018




2 Shandong Oriental Ocean Sci-Tech Co., Ltd., Yantai 264003, China;
3 Key Laboratory of Marine Genetics and Breeding of Ministry of Education, Ocean University of China, Qingdao 266003, China;
4 Institutes of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, China
As an economically important kelp, Saccharina japonica (Laminariales) plays an irreplaceable role in marine ecosystems (Tonon et al., 2011). It has become the largest biomass harvested among all seaweeds because of its extensive cultivation in East Asia, Europe and North America for its three major carbohydrates, alginate, laminarian and mannitol (Nyvall et al., 2003; Takeda et al., 2011; Kraan, 2013). Kelp has also become ideal feedstocks for the production of bioethanol, and their cultivation does not require arable land, fertilizer or fresh water (Wargacki et al., 2012; Enquist-Newman et al., 2014; Yang and Li, 2014). According to the 2016 China Fishery Statistics Yearbook, the annual S. japonica production of China in 2015 reached 2.089 2 million tons, accounting for more than 90% of the world total. Despite the long history of cultivation, we currently have a very limited knowledge of its metabolism.
Saccharina japonica is cultivated upside down under a floating rope net with the holdfast and meristem facing the sunlight and with the blade in the shade, making S. japonica sporophyte persistently experience adverse stresses during its development. Environmental shifts including constantly aggravating pollution, continuously changing climate and runaway intensification of aquaculture scale may interact and worsen these stresses (Ye et al., 2015). The life cycle of S. japonica is alternative between heteromorphic sporophyte and gametophyte. This study focused on the sporophyte because it is cultivated and harvested. The sporophyte of S. japonica can be divided into a series of stages different in morphology and physiology (Wu et al., 2015). The first is mushroom stage at which S. japonica is thin and tender and about one meter long; the second is adult stage at which the blade becomes hard and tough, and the content of organic matter increases significantly; the third is mature stage at which S. japonica stops to grow and sporangia develops on blade surface; and the last is aging stage at which the blade dies gradually. The S. japonica sporophyte changes in morphology and physiology at different developmental stages; however, the physiological mechanisms underlining these changes are still unknown, and the metabolism processes of S. japonica during its development are rarely documented.
With the popularization of massively parallel sequencing technologies, RNA sequencing (RNAseq) has been used to decipher the molecular mechanisms that underline the physiological processes of diverse organisms. The aim of this study was to disclose the difference of expressed genes and the metabolic pathways they defined at different developmental stages of S. japonica through RNAseq.
2 MATERIAL AND METHOD 2.1 Content determination of chemicalsSporophytes were collected from Sanggou Bay, Rongcheng, Shandong Province in China. The sporophytes were cultivated on a rope net floating permanently at the surface of the seawater. The rope net consisted of parallel master ropes anchored to the seafloor and sporophyte hanging ropes knotted between the master ropes at an equal distance. Each sporophyte hanging ropes had the sporophytes of Dongfang No. 7, a variety of S. japonica (Li et al., 2016). At four different developmental stages (mushroom, adult, mature and aging) corresponding to four different months (March, April, June and July, 2015, respectively), sporophytes were collected from three points along a sporophyte hanging rope, each at surface, middle and bottom positions and three sporophyte hanging ropes each stage. In total, 36 sporophytes were collected, which were sun-dried, ground to powders ≥425 μm, mixed, sealed into a plastic bag and stored in dry air and at room temperature. Alginate content was determined with the method described by Shang et al. (2011) while mannitol and iodine contents were assayed following the method described early (Ji, 2004). Simultaneously, the blade length, width, color and flexibility were measured in field and recorded as described early (Li et al., 2016).
2.2 RNA extraction and transcriptome sequencingSporophytes were simultaneously collected with those for determining alginate, mannitol and iodine contents, each at surface, middle and bottom position of a hang rope and two hanging ropes each stage. In total, 24 sporophytes were collected, six each stage. Tissues from the growing point each sporophyte of the same developmental stage were cut, grouped, fast frozen in liquid nitrogen and used to extracting total RNA with TransZol Plant, a kit of Invitrogen, following manufacturer's instructions. The concentration and purity of RNA was checked on the NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, USA). The quality of RNA was determined through electrophoresis in 1% agarose gel in order to ensure that the extracted RNA was not degraded and contaminated by genomic DNA. The mRNA was purified using poly-T attached magnetic beads, fragmented and used as the templates of the firststrand cDNA synthesis using random hexamer primers and M-MuLV reverse transcriptase (RNase H free). The second-strand cDNA was synthesized, subsequently, using RNase H and DNA polymerase I. The double stranded cDNA was end polished with exonuclease and polymerase. After nucleotide "A" addition at the 3′ ends of cDNA fragments, NEBNext Adaptor was linked on. The cDNA in different lengths were selected using AMPure XP system (Beckman Coulter, Beverly, USA) and amplified through PCR, which were purified using AMPure XP system (Beckman Coulter, Beverly, USA), yielding the cDNA library for sequencing. In total, 8 libraries, 2 each stage, were sequenced on an Illumina HiSeqTM2000/-MiSeqTM platform (Novogene Bioinformatics Technology Co., Ltd.).
2.3 Sequence data processingThe sequencing records were translated into the raw reads in FASTA format through CASAVA Base Calling (Zhang et al., 2015; Miao et al., 2016). Clean reads were obtained after removing those either containing adaptor or ploy-N or at low quality. Clean reads were assembled using Trinity (Grabherr et al., 2011) to generate unigenes (here after genes). Differential expression analysis was performed using the DEGseqR package (1.12.0) (Anders and Huber, 2010). The abundance of gene transcripts were estimated based on FPKM (fragment per kilobase of exon length per million reads) values and the abundance of gene transcripts was considered to be significantly differential if q-value < 0.05 (Trapnell et al., 2010). Differentially expressed genes (DEGs) were selected by comparing two adjacent stages of S. japonica development. GO enrichment of DEGs was carried out using the GOseqR Package based on a hypergeometric test (Young et al., 2010). GO functional classifications were performed with WeGO software (Ye et al., 2006). DEGs were also enriched into KEGG pathways (http://www.genome.jp/kegg/) with KOBAS software (Mao et al., 2005). The enrichment P-values were adjusted with Benjamin and Hochberg method. In order to appropriately describe the function of expressed genes, their corresponding models in the S. japonica genome (Ye et al., 2015) were Blast screened against SwissProt for their functional homologs. In addition, the genes involved in sugar biosynthesis were screened out through blasting the expressed gene assemblage with the enzyme amino acid sequences retrieved from NCBI as queries with BLAST v 2.2.9 (Altschul et al., 1997).
3 RESULT AND DISCUSSION 3.1 Morphological and metabolite shifts at different development stagesThe morphological changes of S. japonica at different sporophyte developmental stages mainly manifested in the length, color and flexibility of blade (Fig. 1). The length of blade increased from mushroom to adult, and simultaneously the color became deeper and deeper and the flexibility became tougher and tougher. The most protrusive morphological changes included the blade thickening from mushroom to adult stages and the appearance of sporangia from adult to mature stages. A large number of sporangia protruded from surface of the entire blade at mature stage and were observable with naked eyes at both mature and aging stages. The percentile content of alginate was found to be similar among developmental stages while that of mannitol showed a gradual increase from early to latter developmental stages and that of iodine showed a difference before adult and after mature stages (Table 1).

|
| Fig.1 Morphological differences of sporophyte among mushroom (a), adult (b), mature (c) and aging (d) stages a. light brown and smooth; b. deep brown; c. yellow sporangium observable; d. seeable loss of tissue. c1: a magnified area with sporangia. |

|
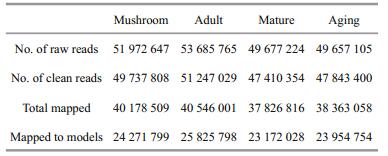
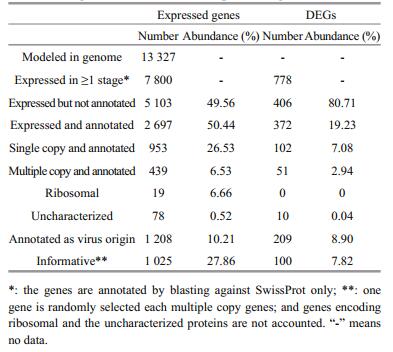
In total, 204 992 741 raw reads were obtained for 4 developmental stages, of them, 196 238 591 were clean, accounting for 95.73% of the total. Of the clean reads, 79.96% were mapped onto the S. japonica genome (Ye et al., 2015), and 49.55% to the gene models of the genome. From the clean reads, 7 800 gene models were found to be transcribed, accounting for 58.53% (7 800/13 327). In total, 915 genes were found to be differentially expressed between stages, of them, 64 were between mushroom and adult stages (27 up-regulated and 37 down-regulated), 539 between adult and mature stages (348 up-regulated and 191 down-regulated), and 302 between mature and aging stages (99 up-regulated and 203 downregulated) (Table 2, Fig. 2).

|
| Fig.2 The histogram of the number of DEGs showing DEGs between S. japonica sporophyte developmental stages (a) and the hot map showing transcript abundance difference between stages and clusters of DEGs (left) and stages (above) (b) There are 64 DEGs between mushroom and adult stages, 539 between adult and mature stages and 302 between mature and aging stages. In (b), the red color represents up-regulation of gene expression while the blue color represents the down-regulation of gene expression, and the deepness of color indicates the degree of abundance change. |
Against SwissProt, only 2 697 genes of 7 800 expressed in total were annotatable. Against more data bases, the number of annotatable genes should be increasable; however, an obvious boost of such number was not expected. We found that the total abundance of the annotatable genes accounted for 50.44% of the total. Of the expressed, we found 778 genes differentially expressed between at least two stages. Of these differentially expressed genes (DEGs), only 372 were annotatable, which accounted 19.23% of the total abundance of DEGs (Table 3).
|
It should be noticed that S. japonica was not a model organism. It is not close to the plant lineage consisting of red and green algae and high plants but rather phylogenetically close to diatoms, dinoflagellates, ciliates among others (Keeling et al., 2005; Baldauf, 2008; Lane and Archibald, 2008). Till present, the genes isolated and characterized in S. japonica were very limited. As a brown algal model, the genome of Ectocarpus siliculosus has been sequenced (Cock et al., 2010), however, a large percentage of genes remained non-annotated. In addition, the genetic and physiological difference between S. japonica and E. siliculosus should be large although they are all brown large. Such scenario may have restricted the annotation of S. japonica genes and also description of its physiological processes, thus it was understandable that less genes were annotatable, especially when the annotation is against only one data base (here SwissProt).
3.3 Weird transcriptsVery interestingly, we found that 1 208 of the 7 800 expressed and 2 697 annotated were virus associating genes and the total abundance of their transcripts accounted for about 10.21% of the total expressed. Such weirdness was observed also in the DEGs. Of 778 DEGs, 372 were annotatable and 209 were virus associating genes with total abundance of their transcripts accounted for 8.90%. It was true that the expressed genes were annotated against SwissProt only, and some of virus associating genes may be functionally assignable against other data bases. However, such a large portion of virus associating genes indicated that the S. japonica genome contained a large portion of virus originating genes, and these genes were active indeed in the current evolved genome. This observation implied that the S. japonica genome was weird. Delaroque and Boland (2008) found that the genome of the brown alga E. siliculosus contains a series of viral DNA pieces, suggesting an ancient association with large dsDNA viruses. It seemed that S. japonica genome did contain also a large portion of virus originating genes, thus evolving in a specific route, which is worthy of further study.
We identified these DESs by referring to the published genome sequence (Ye et al., 2015). Our findings generated multiple, magic and mystery stories to be unveiled. It was obvious that the S. japonica genome should be resequenced with newly available tools and strategies (Eid et al., 2009; Lieberman-Aiden et al., 2009; Burton et al., 2013; Vanburen et al., 2015) to meet the standards of the reference in order to solve the questions such as less annotated genes (Table 3) and possibly missed gene models. Similarly the genes implying S. japonica genome evolution included a large set of functionally repetitive genes. We found that 439 of 2 697 annotated genes were multiple copy genes, which either were or contained the domain of known proteins. Again these genes were worthy of further inspection of their structures and functions (Table 3).
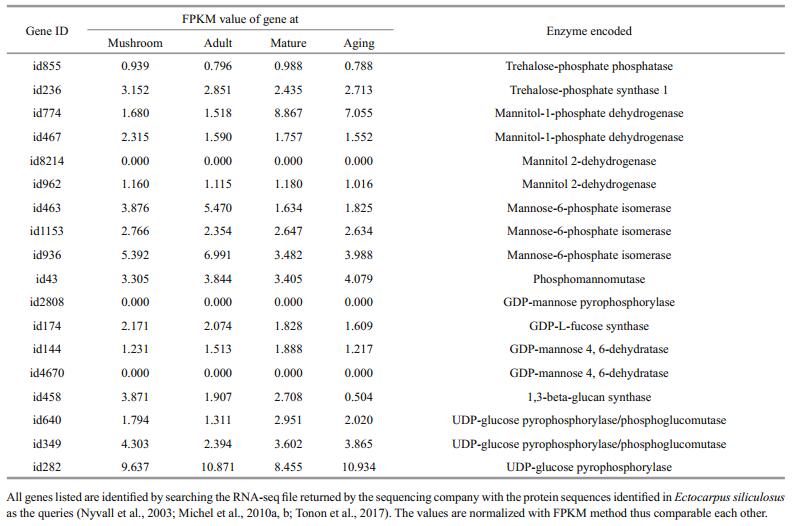
3.4 Gene expression pattern associating with structural and storage sugar changesIn order to describe the physiological mechanisms underlining the morphological and biochemical changes at different developmental stages, we tried to enrich the genes differentially expressed between at least 2 stages, unfortunately, we failed to find any metabolism and signaling pathway. As was pointed out above, the annotated transcripts were less than the expected in this non-model brown alga, and those annotated against SwissProt covered a large portion of virus genes or virus protein domain fusing genes. As an alternative strategy, we turned to find out the genes controlling the biosynthesis of structural and storage sugars, which were identified in recent years from the genome of E. siliculosus (Michel et al., 2010a, b; Tonon et al., 2017) using a local BLAST approach and the previously identified genes as queries. In total, 19 genes were found, which controlled the biosynthesis of 5 types of sugars including sulfated fucan, alginate, mannitol, trehalose and laminarin (Table 4; Fig. 3).
|

|
| Fig.3 The expression patterns of the enzyme encoding genes functioning in the biosynthesis of S. japonica structural and storage sugars The left were biosynthesis pathways of the five sugars and the right were the abundance of the genes involving in these pathways. From green to red, the abundance increases gradually. The abundance is expressed as the FPKM value. |
A FPKM value represents the normalized abundance of a gene transcript. In order to describe the biosynthesis intensity of a sugar, the transcript abundances of the genes involved in its biosynthesis were summed up first in the direction of developmental stages and then in the direction of the list of all genes involved in its biosynthesis with the total sum averaged among the genes involved in its biosynthesis as the intensity of its biosynthesis. Following the same philosophy, the abundances of all genes were summed up in both directions and averaged as the whole genome transcription (background) and used as the reference to the biosynthesis intensity of a sugar. It was found that the transcripts abundance associating with sugar biosynthesis was about 2.13 folds of all expressed genes (10.434/4.893), indicating that the biosyntheses of structural and storage polysaccharides were very important cellular processes of S. japonica. Interestingly, the abundance of genes responsible for structural (alginate) and storage (mannitol and laminarin) polysaccharides was similar to each other (11.538/12.741). From the averaged sum of the abundance of genes involved in the biosynthesis of each polysaccharide, we found that the biosynthesis of laminarin was the strongest process (17.781), which was followed by that of alginate (11.538), mannitol (7.701), trehalose (7.331) and sulfated fucan (4.510). At different developmental stages, the total abundance of genes involved in the biosynthesis of alginate and laminarin was similar among stages, however, that of genes involved in the biosynthesis of mannitol increased about 2 folds from mushroom and adult stages to mature and aging stages. Such trend explained our observations that the content of alginate was almost constant at different developmental stages while that of mannitol increased gradually (Table 1).
3.5 Persistent expression of defense associating genesOf 2 697 genes annotated against SwissProt, we found that a set of defense and cell recurring genes highly expressed in S. japonica sporophyte development, and many of them expressed differentially among stages, which included those encoding vanadium-dependent bromoperoxidase, heat shock 70 protein, and peroxiredoxin and peptide methionine sulfoxide reductase (Table 5). On average, the sum abundance of the transcripts of these genes at 4 developmental stages was 3.40 (24.258/7.138) and 4.96 (24.258/4.893) folds of all annotated and all expressed genes, respectively, indicating that S. japonica sporophytes persistently respond possible pathogen and environmental stresses.

|
Such physiological status is possible as strong UV irradiation, worse pollution, changing climate, intensive cultivation among others may have been making S. japonica sporophytes face to continuous stresses.
The pathways of phytohormone biosynthesis in higher plants have been well described, and the role of phytohormones in the regulation of metabolic processes in algae is no longer in doubt (Kiseleva et al., 2012); however, the hormone metabolism at gene level in different groups of algae remains largely unknown although all known phytohormones are found in various algae, and auxin metabolism and function seem to be characterized in E. siliculosus (Le Bail et al., 2010). Similarly, oxylipins, a group of oxygenized polyunsaturated fatty acids (PUFAs) and derivatives, have been described in diverse algae, especially in brown algae like S. japonica. The mechanism of polyunsaturated fatty acids (PUFAs) oxidation at least via a lipoxygenase step and then alterative and diverse subsequent reactions collectively is referred to oxylipin pathway (Andreou et al., 2009). The biosynthesis of diverse oxylipin is the response of organisms to both abiotic and biotic stresses, which forms a few rings along a chain from the recognition and reception of stress factors and pathogenic initiators and effectors to the transcriptional reprogramming of genome, namely oxylipin signaling, a widely observed response mechanism of organism also referred to the innate immunity (Ritter et al., 2014). As the intensively cultivated kelp species, S. japonica is facing a set of biotic and abiotic stresses, including continuously raising seawater temperature, content of carbon dioxide, strong fluctuation of sunshine, strengthening UV irradiation, territorial sources of metal pollutants, intensifying pathogens due to continuous and high density cultivating among others. The genome sequence S. japonica is available currently (Ye et al., 2015), however the gene assemblage encoding the enzymes functioning in oxylipin synthesis and signaling have not been well characterized yet. The analyses in a model brown alga, E. siliculosus, have shown that this brown alga genome contains neither jasmonate (a group of oxylipins) receptor nor its synthesizing enzyme encoding genes (Cock et al., 2010; Ritter et al., 2014). We speculated that S. japonica may do so because they are closely related each other. It has been hypothesized that S. japonica responds to both abiotic and biotic stresses by up-taking and metabolizing iodine, bursting H2O2, synthesizing both octadecanoids and eicosanoids derived oxylipins, and reprogramming gene transcription of its genome with unknown transcriptional networks (Küpper et al., 1998; Cosse et al., 2009; Ritter et al., 2014; Thomas et al., 2014; Tsuda and Somssich, 2015).
Vanadium-dependent bromoperoxidase is a radiative oxygen species (ROS) detoxifying enzyme specific for brown algae. Heat shock 70 may facilitate the refolding of damaged proteins and aid to protecting protein aggregation under oxidative stress. The other two proteins associate also with the stress response. In fact, these genes were picked up from those annotated against only SwissProt. We are sure that homologous searching should find more stress responding genes. However, our findings should have implied that cultivated S. japonica persistently responds to abiotic and biotic stresses. In recent years, environmental changes may have placed the cultivated S. japonica at a threshold, a little lower making it develop healthily, and a little higher causing the outbreak of diseases. Breeding high disease resistant varieties is appreciated while reforming S. japonica cultivating system may realize such purpose fast.
4 CONCLUSIONTranscripts of kelp (Saccharina japonica) at different sporophyte developmental stages were a little weird. The transcription of associating genes and the metabolite shift of sugars at were consistent with each other. The content of alginate was almost constant at different developmental stages while that of mannitol increased sharply. S. japonica sporophytes persistently respond possible pathogen and environmental stresses by expressing defense genes. Further research is required to reveal the mechanism of S. japonica defense.
5 DATA AVAILABILITY STATEMENTThe data that support the findings of this study are available from the corresponding author on request.
Altschul S F, Madden T L, Schaffer A A, Zhang J H, Zhang Z, Miller W, Lipman D J. 1997. Gapped BLAST and PSIBLAST: A new generation of protein database search programs. Nucleic Acids Research, 25(17): 3389-3402.
DOI:10.1093/nar/25.17.3389 |
Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biology, 11(10): R106.
DOI:10.1186/gb-2010-11-10-r106 |
Andreou A, Brodhun F, Feussner I. 2009. Biosynthesis of oxylipins in non-mammals. Progress in Lipid Research, 48(3-4): 148-170.
DOI:10.1016/j.plipres.2009.02.002 |
Baldauf S L. 2008. An overview of the phylogeny and diversity of eukaryotes. Journal of Systematics and Evolution, 46(3): 263-273.
|
Burton J N, Adey A, Patwardhan R P, Qiu R L, Kitzman J O, Shendure J. 2013. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology, 31(12): 1119-1125.
DOI:10.1038/nbt.2727 |
Cock J M, Sterck L, Rouzé P, Scornet D, Allen A E, Amoutzias G, Anthouard V, Artiguenave F, Aury J M, Badger J H, Beszteri B, Billiau K, Bonnet E, Bothwell J H, Bowler C, Boyen C, Brownlee C, Carrano C J, Charrier B, Cho G Y, Coelho S M, Collén J, Corre E, Da Silva C, Delage L, Delaroque N, Dittami S M, Doulbeau S, Elias M, Farnham G, Gachon C M, Gschloessl B, Heesch S, Jabbari K, Jubin C, Kawai H, Kimura K, Kloareg B, Küpper F C, Lang D, Le Bail A, Leblanc C, Lerouge P, Lohr M, Lopez P J, Martens C, Maumus F, Michel G, Miranda-Saavedra D, Morales J, Moreau H, Motomura T, Nagasato C, Napoli C A, Nelson D R, Nyvall-Collén P, Peters A F, Pommier C, Potin P, Poulain J, Quesneville H, Read B, Rensing S A, Ritter A, Rousvoal S, Samanta M, Samson G, Schroeder D C, Ségurens B, Strittmatter M, Tonon T, Tregear J W, Valentin K, von Dassow P, Yamagishi T, Van de Peer Y, Wincker P. 2010. The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature, 465(7298): 617-621.
DOI:10.1038/nature09016 |
Cosse A, Potin P, Leblanc C. 2009. Patterns of gene expression induced by oligoguluronates reveal conserved and environment-specific molecular defense responses in the brown alga Laminaria digitata. New Phytologist, 182(1): 239-250.
DOI:10.1111/j.1469-8137.2008.02745.x |
Delaroque N, Boland W. 2008. The genome of the brown alga Ectocarpus siliculosus contains a series of viral DNA pieces, suggesting an ancient association with large dsDNA viruses. BMC Evolutionary Biology, 8: 110.
DOI:10.1186/1471-2148-8-110 |
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, de Winter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. 2009. Real-time DNA sequencing from single polymerase molecules. Science, 323(5910): 133-138.
DOI:10.1126/science.1162986 |
Enquist-Newman M, Faust A M E, Bravo D D, Santos C N S, Raisner R M, Hane A, Sarvabhowman P, Le C, Regitsky D D, Cooper S R, Peereboom L, Clark A, Martinez Y, Goldsmith J, Cho M Y, Donohoue P D, Luo L, Lamberson B, Tamrakar P, Kim E J, Villari J L, Gill A, Tripathi S A, Karamchedu P, Paredes C J, Rajgarhia V, Kotlar H K, Bailey R B, Miller D J, Ohler N L, Swimmer C, Yoshikuni Y. 2014. Efficient ethanol production from brown macroalgae sugars by a synthetic yeast platform. Nature, 505(7482): 239-243.
DOI:10.1038/nature12771 |
Grabherr M G, Haas B J, Yassour M, Levin J Z, Thompson D A, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q D, Chen Z H, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren B W, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnology, 29(7): 644-652.
DOI:10.1038/nbt.1883 |
Ji M. 2004. Algal Chemistry. Science Press, Beijing. 812p.
(in Chinese)
|
Keeling P J, Burger G, Durnford D G, Lang B F, Lee R W, Pearlman R E, Roger A J, Gray M W. 2005. The tree of eukaryotes. Trends in Ecology and Evolution, 20(12): 670-676.
DOI:10.1016/j.tree.2005.09.005 |
Kiseleva A A, Tarachovskaya E A, Shishova M F. 2012. Biosynthesis of phytohormones in algae. Russian Journal of Plant Physiology, 59(5): 595-610.
DOI:10.1134/S1021443712050081 |
Kraan S. 2013. Mass-cultivation of carbohydrate rich macroalgae, a possible solution for sustainable biofuel production. Mitigation and Adaptation Strategies for Global Change, 18(1): 27-46.
DOI:10.1007/s11027-010-9275-5 |
Küpper F C, Schweigert N, Gall E A, Legendre J M, Vilter H, Kloareg B. 1998. Iodine uptake in Laminariales involves extracellular, haloperoxidase-mediated oxidation of iodide. Planta, 207(2): 163-171.
DOI:10.1007/s004250050469 |
Lane C E, Archibald J M. 2008. The eukaryotic tree of life: endosymbiosis takes its TOL. Trends in Ecology and Evolution, 23(5): 268-275.
DOI:10.1016/j.tree.2008.02.004 |
Le Bail A, Billoud B, Kowalczyk N, Kowalczyk M, Gicquel M, Le Panse S, Stewart S, Scornet D, Cock J M, Ljung K, Charrier B. 2010. Auxin metabolism and function in the multicellular brown alga Ectocarpus siliculosus. Plant Physiology, 153(1): 128-144.
DOI:10.1104/pp.109.149708 |
Li X J, Zhang Z Z, Qu S C, Liang G J, Sun J, Zhao N, Cui C J, Cao Z M, Li Y, Pan J H, Yu S H, Wang Q Y, Li X, Luo S J, Song S F, Guo L, Yang G P. 2016. Improving seedless kelp (Saccharina japonica) during its domestication by hybridizing gametophytes and seedling-raising from sporophytes. Scientific Reports, 6: 21255.
DOI:10.1038/srep21255 |
Lieberman-Aiden E, van Berkum N L, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie B R, Sabo P J, Dorschner M O, Sandstrom R, Bernstein B, Bender M A, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny L A, Lander E S, Dekker J. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326(5950): 289-293.
DOI:10.1126/science.1181369 |
Mao X, Cai T, Olyarchuk J G, Wei L. 2005. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics, 21(19): 3787-3793.
DOI:10.1093/bioinformatics/bti430 |
Miao Y Y, Zhu Z B, Guo Q S, Zhu Y H, Yang X H, Sun Y. 2016. Transcriptome analysis of differentially expressed genes provides insight into stolon formation in Tulipa edulis. Frontiers in Plant Science, 7: 409.
|
Michel G, Tonon T, Scornet D, Cock J M, Kloareg B. 2010a. Central and storage carbon metabolism of the brown alga Ectocarpus siliculosus: Insights into the origin and evolution of storage carbohydrates in Eukaryotes. New Phytologist, 188(1): 67-81.
DOI:10.1111/j.1469-8137.2010.03345.x |
Michel G, Tonon T, Scornet D, Cock J M, Kloareg B. 2010b. The cell wall polysaccharide metabolism of the brown alga Ectocarpus siliculosus: Insights into the evolution of extracellular matrix polysaccharides in Eukaryotes. New Phytologist, 188(1): 82-97.
DOI:10.1111/j.1469-8137.2010.03374.x |
Nyvall P, Corre E, Boisset C, Barbeyron T, Rousvoal S, Scornet D, Kloareg B, Boyen C. 2003. Characterization of mannuronan C-5-epimerase genes from the brown alga Laminaria digitata. Plant Physiology, 133(2): 726-735.
DOI:10.1104/pp.103.025981 |
Ritter A, Dittami S M, Goulitquer S, Correa J A, Boyen C, Potin P, Tonon T. 2014. Transcriptomic and metabolomic analysis of copper stress acclimation in Ectocarpus siliculosus highlights signaling and tolerance mechanisms in brown algae. BMC Plant Biology, 14: 116.
DOI:10.1186/1471-2229-14-116 |
Shang D R, Ning J S, Zhao Y F, Zhai Y X, Shu B S, Sheng X F, Guo Y Y. 2011. Establishment of the determination on kelp alginate. Food Science and Technology, 36(8): 252-254.
(in Chinese with English abstract) |
Takeda H, Yoneyama F, Kawai S, Hashimoto W, Murata K. 2011. Bioethanol production from marine biomass alginate by metabolically engineered bacteria. Energy & Environmental Science, 4(7): 2575-2581.
|
Thomas F, Cosse A, Le Panse S, Kloareg B, Potin P, Leblanc C. 2014. Kelps feature systemic defense responses: Insights into the evolution of innate immunity in multicellular eukaryotes. New Phytologist, 204(3): 567-576.
DOI:10.1111/nph.12925 |
Tonon T, Eveillard D, Prigent S, Bourdon J, Potin P, Boyen C, Siegel A. 2011. Toward systems biology in brown algae to explore acclimation and adaptation to the shore environment. Omics, 15(12): 883-892.
DOI:10.1089/omi.2011.0089 |
Tonon T, Li Y, McQueen-Mason M. 2017. Mannitol biosynthesis in algae: More widespread and diverse than previously thought. New Phytologist, 213(4): 1573-1579.
DOI:10.1111/nph.2017.213.issue-4 |
Trapnell C, Williams B A, Pertea G, Mortazavi A, Kwan G, van Baren M J, Salzberg S L, Wold B J, Pachter L. 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28(5): 511-515.
DOI:10.1038/nbt.1621 |
Tsuda K, Somssich I E. 2015. Transcriptional networks in plant immunity. New Phytologist, 206(3): 932-947.
DOI:10.1111/nph.2015.206.issue-3 |
Vanburen R, Bryant D, Edger P P, Tang H B, Burgess D, Challabathula D, Spittle K, Hall R, Gu J, Lyons E, Freeling M, Bartels D, Ten Hallers B, Hastie A, Michael T P, Mockler T C. 2015. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature, 527(7579): 508-511.
DOI:10.1038/nature15714 |
Wargacki A J, Leonard J F, Win M N, Regitsky D D, Santos C N S, Kim P B, Cooper S R, Raisner R M, Herman A, Sivitz A B, Lakshmanaswamy A, Kashiyama Y, Baker D, Yoshikuni Y. 2012. An Engineered microbial platform for direct biofuel production from brown macroalgae. Science, 335(6066): 308-313.
DOI:10.1126/science.1214547 |
Wu H Y, Wang X Y, Zhu A C. 2015. Analysis of component changes of Saccharina japonica at mushroom-adult stage. Marine Sciences, 39(8): 35-38.
(in Chinese with English abstract) |
Yang G P, Li F F. 2014. Fermenting brown algal carbohydrates into ethanol with engineered microorganisms: a new horizon for bioethanol production. Marine Sciences, 38(4): 88-95.
(in Chinese with English abstract) |
Ye J, Fang L, Zheng H K, Zhang Y, Chen J, Zhang Z J, Wang J, Li S T, Li R Q, Bolund L, Wang J. 2006. WEGO: a web tool for plotting GO annotations. Nucleic Acids Research, 34(S2): W293-W297.
|
Ye N H, Zhang X W, Miao M, Fan X, Zheng Y, Xu D, Wang J F, Zhou L, Wang D S, Gao Y, Wang Y T, Shi W Y, Ji P F, Li D M, Guan Z, Shao C W, Zhuang Z M, Gao Z Q, Qi J, Zhao F Q. 2015. Saccharina genomes provide novel insight into kelp biology. Nature Communications, 6: 6986.
DOI:10.1038/ncomms7986 |
Young M D, Wakefield M J, Smyth G K, Oshlack A. 2010. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biology, 11(2): R14.
DOI:10.1186/gb-2010-11-2-r14 |
Zhang Y H, Li W J, Dou Y J, Zhang J X, Jiang G H, Miao L X, Han G F, Liu Y X, Li H, Zhang Z H. 2015. Transcript quantification by RNA-seq reveals differentially expressed genes in the red and yellow fruits of Fragaria vesca. PLoS One, 10(12): e0144356.
DOI:10.1371/journal.pone.0144356 |