 2019, Vol. 37
2019, Vol. 37Institute of Oceanology, Chinese Academy of Sciences
Article Information
- YU Weiting, ZHANG Demeng, LIU Xiudong, WANG Yunhong, TONG Jun, ZHANG Mengxue, MA Xiaojun
- Amphiphilic sodium alginate-vinyl acetate microparticles for drug delivery
- Journal of Oceanology and Limnology, 37(3): 855-862
- http://dx.doi.org/10.1007/s00343-019-8127-8
Article History
- Received May. 9, 2018
- accepted in principle Jun. 20, 2018
- accepted for publication Nov. 4, 2018




2 Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China;
3 College of Environment and Chemical Engineering, Dalian University, Dalian Economic Technological Development Zone, Dalian 116622, China;
4 State Key Laboratory of Bioactive Seaweed Substances, Qingdao Brightmoon Seaweed Group Co. Ltd., Qingdao 266400, China;
5 Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
Alginate is a naturally derived polysaccharide extracted from brown seaweed. It is a kind of linear anionic polymer with the structural units of β-Dmannuronic acid (M) and α-L-guluronic acid (G) residues. They are randomly linked by 1, 4-glycosidic bond to form homogenous blocks (MM and GG) and heterogeneous blocks (GM) along the polymer chains depending on the source. Most of the commercially available alginates are in the form of sodium alginate (Na-Alg). Specifically, sodium alginate solution can transform from sol to hydrogel state under biologically benign conditions (room temperature and near-neutral pH) when divalent cations such as Ca2+ are added to trigger the ionotropy effect between the divalent cations and Na+. The formed calcium alginate (CaAlg) hydrogel has three dimensional (3D) network structure containing more than 90% of water, which is thought to be beneficial for the entrapment of bioactive substances. Together with its biocompatibility, low toxicity, and relatively low cost, alginate hydrogels have been extensively investigated and used for many biomedical applications including cell transplantation, tissue regeneration and drug delivery (Liu et al., 2008; Lee and Mooney, 2012).
Generally, alginate hydrogels can be fabricated in the form of microparticles or microcapsules by various preparation methods such as electrostatic droplets, emulsification technique (Zhang et al., 2008; Song et al., 2013; Ching et al., 2017; Lopes et al., 2017), which have emerged as a competitive drug delivery platform due to above mentioned advantages. Both small chemical drugs and macromolecular proteins have been loaded in alginate particles (Liakos et al., 2013; Shin et al., 2013; Zhang et al., 2013; Du and Stenzel, 2014), and some alginate-based formulations can release drugs in a controlled manner according to the parameters optimization of materials and technical process. However, the 3D porous network and hydrophilicity of alginate hydrogels (pore size ~5 nm), always led to the rapid release of small molecules through the gel (Boontheekul et al., 2005). Moreover, macromolecular proteins also displayed 'burst release' behavior from chitosan coated alginate hydrogel microcapsules, which resulted in the reduction of drug bioavailability (Desai et al., 2010).
To resolve the problem of fast release, alginate modification via hydrophobic molecules or moieties has been proposed as a strategy to retard the release rate of hydrophilic drugs. The hydroxyl (-OH at C-2 and C-3) and carboxyl groups (-COOH at C-6) along the chain backbone of alginate can be modified for different purposes such as esterification, oxidation, and amidation (Yang et al., 2011; Pawar and Edgar, 2012). Acrylic, acrylamide, and vinyl acetate monomers have been grafted on alginate molecules or alginate hydrogel beads to form copolymers and used as drug carriers (Fang et al., 2005; Xu and Ni, 2012). However, the degree of substitution (DS) of the hydrophobic part on alginate molecules or gel beads was over 100% so that the hydrophobic part became the predominant structure of copolymers. This transformation suggested the solubility and biocompatibility of hydrophilic alginate were changed, which is against its application in drug delivery of bioactive molecules such as proteins. In our previous report, amphiphilic sodium alginatevinyl acetate (Alg-g-PVAc) with a lower degree of substitution (< 30%) was synthesized by homogeneous polymerization (Xu et al., 2013), which can keep the predominant hydrophilic nature of the product and be easily dissolved in the water for further manufacture and application.
With the purpose of retarding the release of macromolecular drugs from hydrophilic alginatebased carriers, hydrophobically modified alginate was synthesized to prepare drug carriers and tested for the release of macromolecular drug model. In detail, alginate modified via hydrophobic molecules (vinyl acetate) was used to firstly fabricate amphiphilic Alg-g-PVAc hydrogel beads by emulsification/internal gelation technique. Bovine serum albumin (BSA) was used as a model drug and entrapped into the amphiphilic hydrogel beads. Then another polysaccharide chitosan was assembled on the surface of beads to form novel BSA-loaded Alg-g-PVAc/chitosan (Alg-g-PVAc/CS) microcapsules. Finally, the physiochemical properties, drug loading and release behavior in simulated gastric fluid and simulated intestinal fluid from amphiphilic Alg-gPVAc/CS microcapsule were evaluated.
2 MATERIAL AND METHOD 2.1 MaterialSodium alginate was purchased from the Qingdao Crystal Rock Biotechnological Development Co., Ltd. (Qingdao, China), whose viscosity is 0.81 Pa·s as labeled. The composition ratio of α-L-guluronic acid (G) residues and β-D-mannuronic acid (M) residues of alginate were characterized by 1H NMR as G/M ratio of 34/66. Vinyl acetate was analytical reagent from Sinopharm Group. BSA (Mw=6.6 kDa), Tween 80 and Span 80 were purchased from Chemical Reagents Co. Ltd. (Sinopharm Group). Potassium persulfate, liquid paraffin, and absolute ethanol were analytical reagents from Tianjin Damao Chemical Reagent Factory. Chitosan (deacetylation degree of 96%, Mw 50, 150, and 250 kDa) was degraded from raw chitosan (Yuhuan Ocean Biomaterials Corporation, China) with gamma (γ) rays irradiation by Key Laboratory of Nuclear Analysis Techniques, Chinese Academy of Sciences. All other reagents and solvents were of reagent grade and were used without further purification.
2.2 Preparation of BSA-loaded amphiphilic Alg-gPVAc/CS microcapsulesAmphiphilic sodium alginate-vinyl acetate (Alg-gPVAc) synthesized by homogeneous polymerization was dissolved in deionized water in a concentration of 1.5% w/v. 0.1 g BSA and 0.1 g CaCO3 were added under stir state to form a uniform suspension. Then, the suspension was dispersed into liquid paraffin containing 0.5% (v/v) Span 80 for emulsification according to our established protocol (Song et al., 2013). After emulsification, the addition of glacial acetic acid dissociated CaCO3 and released Ca2+ to trigger the gelation. The BSA-loaded calcium Alg-gPVAc gel beads were collected and successively rinsed with 1% (v/v) Tween 80 solution quickly. At last, the BSA-loaded calcium Alg-g-PVAc gel beads were further immersed in 0.5% (w/v) chitosan solution (0.1 mol/L acetate buffer) at the beads/solution ratio of 1:10 to form BSA-loaded Alg-gPVAc/CS microcapsules. Meantime, hydrophilic alginate was used to prepare BSA-loaded alginate/chitosan (AC) microcapsules with the same procedure as a control.
2.3 Morphology and size of BSA-loaded amphiphilic Alg-g-PVAc/CS microcapsulesSome droplets containing BSA-loaded Alg-gPVAc/CS microcapsules were transferred on a watch glass and observed with an XDS-1C Inverted Research Microscope (Chongqing COIC instrument Co. Ltd., China) at a magnification of 100×. Meantime, the size of Alg-g-PVAc/CS microcapsules was determined with laser particle analyzer (LA-960, Horiba Group, Japan).
2.4 Determination of drug loading and loading efficiencyFirstly, the standard curve of absorbance (A)-BSA concentration (C) was obtained by UV-VIS spectrophotometer (UV-2600, Shimadzu Co. Japan) at the maximum absorption wavelength of 278.5 nm. Then, BSA-loaded microcapsules were added into sodium citrate solution (1:10 v/v), and the mixture was vibrated for sometimes to completely disrupt microcapsules. The absorbance of the supernatant was measured at 278.5 nm and the released BSA was calculated with a standard curve. Finally, the drug loading and loading efficiency were calculated according to the following equations.
For the drug loading (DL):
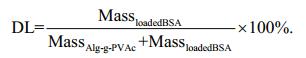 (1)
(1)For the loading efficiency (LE):
 (2)
(2)Simulated gastric fluid (SGF): the pepsin was dissolved in 0.2% NaCl (w/v) solution to reach a final concentration of 0.3 g/L, and then pH was adjusted to 1.0 with concentrated hydrochloric acid.
Simulated intestinal fluid (SIF): The trypsin and bile salt were dissolved in phosphate buffer (0.1 mol/L PBS, pH=7.4) to reach a final concentration of 1.0 g/L and 4.5 g/L, respectively.
An amount of 10-mL BSA-loaded amphiphilic Alg-g-PVAc/CS microcapsules was transferred into a beaker with 100 mL of SGF. Then, the beaker was placed in constant temperature incubator shaker (THZ-98 AB, Shanghai Yiheng Scientific Instrument Co., China) and shaken at 120 r/min (37℃). At fixed time interval (2 min, 5 min, 10 min, 15 min, 30 min, 1 h, and 2 h), 5 mL supernatant was taken out and analyzed by UV-VIS spectrophotometer (UV-2600, Shimadzu Co. Japan) at 278.5 nm. At each time, 5-mL fresh SGF was supplemented.
After 2-h release in SGF, BSA-loaded amphiphilic Alg-g-PVAc/CS microcapsules were separated and quickly rinsed with deionized water. Then they were put into a beaker with 100 mL of SIF, which was placed in constant temperature incubator shaker (THZ-98 AB, Shanghai Yiheng Scientific Instrument Co., China) and shaken at 120 r/min (37℃). At fixed time interval (2 min, 5 min, 10 min, 15 min, 30 min, 1 h, 2 h, 3 h, 4 h, 16 h, and 24 h), 5 mL supernatant was taken out and analyzed by UV-VIS spectrophotometer (UV-2600, Shimadzu Co. Japan) at 278.5 nm. At each time, 5 mL fresh SGF was supplemented.
3 RESULT AND DISCUSSION 3.1 Morphology and size of BSA-loaded amphiphilic Alg-g-PVAc/CS microcapsulesAs shown in Fig. 1, the negatively charged Alg-gPVAc is mixed with BSA and prepared as BSA-loaded Alg-g-PVAc gel beads (Section 2.2). Then they are immersed in positively charged chitosan (CS) solution to form the microcapsule with polyelectrolyte complex membrane (as the arrows indicated in Fig. 2). BSAloaded amphiphilic Alg-g-PVAc/CS microcapsules were observed with an inverted microscope. The amphiphilic microcapsules show good sphericity and smooth surface (Fig. 2). Both the optical photograph and the particle curve demonstrate the amphiphilic microcapsules have a relatively uniform size, and the average diameter is 141±32 μm calculated by the software of the particle analyzer. These results indicate the morphology of amphiphilic Alg-g-PVAc/CS microcapsules have no obvious difference to traditional hydrophilic alginate/chitosan microcapsules (Liu et al., 2008), suggesting that the moderately hydrophobic modification on alginate does not affect the gelation and membrane formation process.

|
| Fig.1 The illustration of the formation of Alg-g-PVAc/CS microcapsule |

|
| Fig.2 The photograph (magnification 100×) and particle size distribution of BSA-loaded Alg-g-PVAc/CS microcapsules The arrows indicate the microcapsule membrane. |
By controlling the reaction conditions, amphiphilic sodium alginate-vinyl acetate (Alg-g-PVAc) with different degree of substitution (DS) of hydrophobic moieties was synthesized by homogeneous polymerization (Xu et al., 2013). Here, Alg-g-PVAc samples with DS of 11.06%, 13.75%, and 19.80% were used to prepare BSA-loaded Alg-g-PVAc/CS microcapsules. The drug loading (DL) and loading efficiency (LE) of BSA in the amphiphilic microcapsules at different DS were measured and calculated as shown in Table 1. Meantime, BSAloaded AC microcapsules were used as a control. DL and LE of BSA in Alg-g-PVAc/CS microcapsules are higher than those of BSA in traditional hydrophilic AC microcapsules. Moreover, DL and LE of BSA in Alg-g-PVAc/CS microcapsules slightly increase with the increase of DS. This can be attributed to the graft of polyvinyl acetate (PVAc) on alginate backbone, which increases the hydrophobicity of copolymer. The hydrophobic interaction may occur between the nonpolar chain segments of BSA and PVAc, and it is beneficial to limit more proteins in the amphiphilic microcapsules during preparation process compared to AC microcapsules. Therefore, both DL and LE of BSA increase.

|
BSA-loaded Alg-g-PVAc/CS microcapsules were prepared with DS of 11.06%, 13.75%, and 19.80%, and they were put sequentially into SGF (2 h) and SIF (24 h) to evaluate BSA release behavior. In general, the accumulative release of BSA in SGF is lower than that in SIF (Fig. 3). The release rate of BSA from hydrophilic AC microcapsules is faster than that from amphiphilic Alg-g-PVAc/CS microcapsules. In addition, DS of hydrophobic PVAc on alginate affects the release of BSA from Alg-g-PVAc/CS microcapsules. The accumulative release of BSA reaches about 90%, 80%, 70%, and 60% from AC, Alg-g-PVAc11.06%/CS, Alg-g-PVAc13.75%/CS, and Algg-PVAc19.80%/CS microcapsules, respectively. In other words, the higher the DS of PVAc is, the slower the release rate is.

|
| Fig.3 The release of BSA from amphiphilic Alg-g-PVAc/CS microcapsules with different DS in simulated gastric fluid and simulated intestinal fluid |
It is well known that alginate hydrogel is a pHresponsive carrier (Mei et al., 2014), which can shrink in acidic solution but swell in neutral or alkaline medium. The shrinkage causes the decrease of pore size of alginate hydrogel and benefits for the retardation of substance diffusion. Therefore, the release of BSA is relatively slow in SGF. Moreover, the coating of chitosan on the surface of alginate hydrogel beads forms the microcapsule membrane. In general, the thicker and denser membrane formed by regulating the parameters (such as Mw of chitosan and membrane formation time) can help reduce the pore size of the network and then decrease the release rate of BSA. On the other hand, more hydrophobic moieties (PVAc) grafted on alginate facilitates abovementioned hydrophobic interaction to significantly retard the release of BSA from amphiphilic Alg-gPVAc/CS microcapsules.
3.3.2 Effect of Mw of chitosan on BSA releaseThe purpose of coating chitosan on the surface of alginate hydrogel beads is to form a membrane and to regulate the release of drugs. Here, chitosan samples with different Mw (50 kDa, 150 kDa, and 250 kDa) were used to complex with Alg-g-PVAc (DS 13.75%) beads to form amphiphilic Alg-g-PVAc/CS microcapsules. The release of BSA in SGF and SIF also displays the different trends (Fig. 4). The Mw of chitosan hardly had an impact on the release of BSA in SGF probably due to the strong pH responsive effect of alginate hydrogel. In SIF, however, the accumulative release of BSA from Alg-g-PVAc/CS microcapsules reduces along the decrease of chitosan Mw. Because the smaller Mw of chitosan means the shorter molecular segments, which is easier to diffuse into the network of hydrogel beads, they can provide more positive amino groups to complex with negative carboxyl groups of alginate (Yu et al., 2010). As a result, thicker microcapsule membrane is formed, which effectively retards the outward diffusion rate of BSA. On the contrary, the longer molecular segments with bigger Mw are relatively hard to diffuse into the network of alginate hydrogel and only react with alginate molecules on the bead surface to form a thinner membrane. Therefore, the release of BSA is faster from amphiphilic Alg-g-PVAc/CS microcapsules with higher chitosan Mw.

|
| Fig.4 The release of BSA from amphiphilic Alg-g-PVAc/CS microcapsules with different chitosan Mw in simulated gastric fluid and simulated intestinal fluid |
Chitosan samples (Mw 150 kDa) were assembled on Alg-g-PVAc (DS 13.75%) beads to form amphiphilic Alg-g-PVAc/CS microcapsules at different membrane formation time (10 min, 15 min, and 20 min). The release of BSA in SGF and SIF is shown in Fig. 5. Similarly, the release of BSA in SIF is higher than that in SGF. To prolong the membrane formation time, more chitosan molecules can diffuse into the deeper network of alginate hydrogel beads, and then the complex reaction between chitosan and alginate under electrostatic interaction is sufficient to form a thick membrane. The resistance of the hydrogel network and membrane to BSA diffusion obviously increases, so that the accumulative release of BSA decreases.

|
| Fig.5 Release of BSA from amphiphilic Alg-g-PVAc/CS microcapsules at different membrane formation time in a simulated gastric fluid and simulated intestinal fluid |
In this paper, the hydrophobically modified alginate was used to prepare novel amphiphilic Alg-g-PVAc/CS microcapsules. The hydrophobic modification of alginate has no effect on the formation of hydrogel beads and microcapsules. After entrapping model protein drug (BSA), the amphiphilic Alg-g-PVAc/CS microcapsules displays good sphericity and relatively uniform size. Compared to traditional hydrophilic AC microcapsules, the drug loading (DL) and loading efficiency (LE) of BSA in Alg-g-PVAc/CS microcapsules are higher, and the release rate of BSA from Alg-g-PVAc/CS microcapsules is slower. Moreover, with the increase of degree of substitution of hydrophobic moieties (PVAc) from 11.06% to 19.80%, DL slightly increased by 5.89%, and LE increased by 8.75%, while the accumulative release of BSA from Alg-g-PVAc/CS microcapsules obviously reduces from about 80% (DS 11.06%) to 60% (DS 19.80%). These results demonstrate that the graft of hydrophobic PVAc on alginate molecules can effectively retard BSA release from microcapsules. Furthermore, the regulation of parameters of chitosan (Mw and membrane formation time) can adjust the properties of the microcapsule membrane to postpone the release of BSA.
5 DATA AVAILABILITY STATEMENTAll data generated and/or analyzed during this study are included in this published article. The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Boontheekul T, Kong H J, Mooney D J. 2005. Controlling alginate gel degradation utilizing partial oxidation and bimodal molecular weight distribution. Biomaterials, 26(15): 2455-2465.
DOI:10.1016/j.biomaterials.2004.06.044 |
Ching S H, Bansal N, Bhandari B. 2017. Alginate gel particles—a review of production techniques and physical properties. Critical Reviews in Food Science and Nutrition, 57(6): 1133-1152.
DOI:10.1080/10408398.2014.965773 |
Desai S, Perkins J, Harrison B S, Sankar J. 2010. Understanding release kinetics of biopolymer drug delivery microcapsules for biomedical applications. Materials Science and Engineering: B, 168(1-3): 127-131.
DOI:10.1016/j.mseb.2009.11.006 |
Du A W, Stenzel M H. 2014. Drug carriers for the delivery of therapeutic peptides. Biomacromolecules, 15(4): 1097-1114.
DOI:10.1021/bm500169p |
Fang Y H, Xiao C M, Wu H, Lin S B. 2005. Graft copolymerization of vinyl acetate onto calcium alginate aquagel. Journal of Huaqiao University (Natural Science), 26(2): 138-140.
(in Chinese with English abstract) |
Lee K Y, Mooney D J. 2012. Alginate: properties and biomedical applications. Progress in Polymer Science, 37(1): 106-126.
DOI:10.1016/j.progpolymsci.2011.06.003 |
Liakos I, Rizzello L, Bayer I S, Pompa P P, Cingolani R, Athanassiou A. 2013. Controlled antiseptic release by alginate polymer films and beads. Carbohydrate Polymers, 92(1): 176-183.
DOI:10.1016/j.carbpol.2012.09.034 |
Liu X D, Yu W T, Wang W, Xiong Y, Ma X J, Yuan Q. 2008. Polyelectrolyte microcapsules prepared by alginate and chitosan for biomedical application. Progress in Chemistry, 20(1): 126-139.
(in Chinese with English abstract) |
Lopes M, Abrahim B, Veiga F, Seiça R, Cabral L M, Arnaud P, Andrade J C, Ribeiro A J. 2017. Preparation methods and applications behind alginate-based particles. Expert Opinion on Drug Delivery, 14(6): 769-782.
DOI:10.1080/17425247.2016.1214564 |
Mei L, He F, Zhou R Q, Wu C D, Liang R, Xie R, Ju X J, Wang W, Chu L Y. 2014. Novel intestinal-targeted Ca-alginatebased carrier for pH-responsive protection and release of lactic acid bacteria. ACS Applied Materials & Interfaces, 6(8): 5962-5970.
DOI:10.1021/am501011j |
Pawar S N, Edgar K J. 2012. Alginate derivatization: a review of chemistry, properties and applications. Biomaterials, 33(11): 3279-3305.
DOI:10.1016/j.biomaterials.2012.01.007 |
Shin S H, Lee J, Lim K S, Rhim T, Lee S K, Kim Y H, Lee K Y. 2013. Sequential delivery of TAT-HSP27 and VEGF using microsphere/hydrogel hybrid systems for therapeutic angiogenesis. Journal of Controlled Release, 166(1): 38-45.
DOI:10.1016/j.jconrel.2012.12.020 |
Song H Y, Yu W T, Gao M, Liu X D, Ma X J. 2013. Microencapsulated probiotics using emulsification technique coupled with internal or external gelation process. Carbohydrate Polymers, 96(1): 181-189.
DOI:10.1016/j.carbpol.2013.03.068 |
Xu G Z, Liu X D, Wang B, Yu W T, Ding D H, Ma X J. 2013. Graft copolymerization of sodium alginate-vinyl acetate and performance evaluation. Chemical Research and Application, 25(8): 1097-1101.
(in Chinese with English abstract) |
Xu Y L, Ni C H. 2012. Synthesis of amphiphilic block copolymer via graft polymerization of DAAM and SA. Applied Chemical Industry, 41(2): 278-281.
(in Chinese with English abstract) |
Yang J S, Xie Y J, He W. 2011. Research progress on chemical modification of alginate: a review. Carbohydrate Polymers, 84(1): 33-39.
DOI:10.1016/j.carbpol.2010.11.048 |
Yu W T, Lin J Z, Liu X D, Xie H G, Zhao W, Ma X J. 2010. Quantitative characterization of membrane formation process of alginate-chitosan microcapsules by GPC. Journal of Membrane Science, 346(2): 296-301.
DOI:10.1016/j.memsci.2009.09.049 |
Zhang X L, He H Y, Yen C, Ho W, Lee L J. 2008. A biodegradable, immunoprotective, dual nanoporous capsule for cell-based therapies. Biomaterials, 29(31): 4253-4259.
DOI:10.1016/j.biomaterials.2008.07.032 |
Zhang Y, Chan H F, Leong K W. 2013. Advanced materials and processing for drug delivery: the past and the future. Advanced Drug Delivery Reviews, 65(1): 104-120.
DOI:10.1016/j.addr.2012.10.003 |