 2019, Vol. 37
2019, Vol. 37Institute of Oceanology, Chinese Academy of Sciences
Article Information
- LI Yingjie, CAO Wenrui, WANG Yan, MA Qingjun
- Microbial diversity in the sediments of the southern Mariana Trench
- Journal of Oceanology and Limnology, 37(3): 1024-1029
- http://dx.doi.org/10.1007/s00343-019-8131-z
Article History
- Received May. 2, 2018
- accepted in principle Jul. 2, 2018
- accepted for publication Aug. 5, 2018




2 Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266000, China;
3 Key Laboratory of Marine Geology and Environment, Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China;
4 University of Chinese Academy of Sciences, Beijing 100049, China;
5 Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China
Hadal oceanic trenches, such as the Mariana Trench represent the deepest habitats on the surface of the Earth. It is located in the western equatorial Pacific, extending to a depth of ~11 km (Nakanishi and Hashimoto, 2011). For a long time, this trench environment was considered as biological deserts, and due to the inaccessibility of the hadal regions of the Mariana Trench, the knowledge of its biology, biogeochemistry, and ecology was very limited. Until recent decades, as the development of operating instrumentation and sampling sediments, a wave of hadal investigation has been stimulated (Takami et al., 1997; Kato et al., 1998; Nogi and Kato, 1999; Glud et al., 2013; Nunoura et al., 2015; Luo et al., 2017). This hadal water environment (6 000 –10 257 m) contains a high level of microbial diversity, and the heterotrophic populations were more abundant while the compositions of the chemolithotrophic population were fewer compared to the abyssal compositions of microbial community (below 4 000 m) (Fig. 1, Nunoura et al., 2015). In another site of the hadal regions of the Mariana Trench, the bacterial communities within the bottom water (~1 m above seafloor) were different from other deep-sea microbial populations (Tarn et al., 2016). In sediments, although Glud et al. (2013) speculated that higher microbial activity levels may occur based on the observation of the enhanced deposition of organic matter at Challenger Deep (Glud et al., 2013), little is known about its microbial diversity and its dynamic (Kato et al., 1998; Pathom-aree et al., 2006; Yoshida et al., 2013; León-Zayas et al., 2017). In this study, we extracted and sequenced PCR amplicons for the V3 to V4 hypervariable region of the bacterial 16S rRNA gene from the sediment taken from the southern flank near the southern end of the Mariana Trench (Fig. 1), thereby documenting the compositions of the sediment microbial community in this region.

|
| Fig.1 The study area and the sample locations a. the location of the Challenger Deep (from https://en.wikipedia.org/wiki/Mariana_Trench); b. close-up view of the Challenger Deep R1 (Nunoura et al., 2015), R2 (Tarn et al., 2016), and the sampling site of this study (SC). |
The sediment core is 2 m long, and was collected on board of R/V Dongfanghong No. 2 (September 3 to 29, 2016) at 142°17.325′E/10°21.213′N in 4 500 m depth at 4℃ (SC, Fig. 1). The core was kept at -80℃ for DNA extraction. The sediments at 30, 94, and 151 cm were used, and coded as M30, M94, and M151, respectively.
2.2 DNA extraction and sequencingMicrobial DNA was respectively extracted from sediment samples M30, M94, and M151 using the E.Z.N.A. stool DNA Kit (Omega Biotek, Norcross, GA, U.S.). The 16S rDNA V3-V4 region was amplified by PCR (95℃ for 2 min, followed by 27 cycles at 98℃ for 10 s, 62℃ for 30 s, and 68℃ for 30 s and a final extension at 68℃ for 10 min) using primers 341F: CCTACGGGNGGCWGCAG; 806R: GGACTACHVGGGTATCTAAT, where the barcode is an eight-base sequence unique to each species. PCR reactions were performed in triplicate 50 μL mixture containing 5 μL of 10×KOD Buffer, 5 μL of 2.5 mmol/L dNTPs, 1.5 μL of each primer (5 μmol/L), 1 μL of KOD polymerase, and 100 ng of template DNA.
Amplicons were extracted from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, U.S.) according to the manufacturer's instructions and quantified using QuantiFluor-ST (Promega, U.S.). Purified amplicons were pooled in equimolar and paired-end sequenced (2×250) on an Illumina Hiseq 2500 platform according to the standard protocols. The raw reads were uploaded into the NCBI Sequence Read Archive (SRA) database (Accession Number: PRJNA449582).
2.3 16S rDNA sequence analysisTo obtain high-quality clean reads, raw reads were first filtered based on the following rules: 1) removing reads containing more than 10% of unknown nucleotides (N); 2) removing reads containing less than 80% of bases with quality (Q-value)>20. Then the filtered reads were assembled into tags according to overlap between paired-end reads with more than 10 bp overlap, and less than 2% mismatch. The software MOTHUR v.1.34.0 (Schloss et al., 2009) was employed to remove the redundant tags to get unique tags that were further used to calculate the abundance. Clean tags were searched against the reference database (http://drive5.com/uchime/uchime_download.html) to perform Reference-based chimera checking using UCHIME algorithm (http://www.drive5.com/usearch/manual/uchime_algo.html). All chimeric tags were removed and effective tags were obtained for further analysis.
The effective tags were clustered into operational taxonomic units (OTUs) of ≥ 97% similarity using UPARSE pipeline (Edgar, 2013). The tag sequence with the highest abundance was selected as a reprehensive sequence within each cluster. The representative sequences were classified into organisms by a naive Bayesian model using RDP classifier v. 2.2 (Wang et al., 2007) based on Greengenes Database (https://www.arb-silva.de/, DeSantis et al., 2006). The abundance statistics of each taxonomy and a phylogenetic tree was constructed in a Perl script and visualized using SVG. Biomarker features in each group were screened by Metastats and LEfSe software.
Chao1, Simpson and all other alpha diversity index were calculated using QIIME. OTU rarefaction curve and Rank abundance curves were plotted in QIIME. Statistics of between-group Alpha index comparison was calculated by a Welch's t-test and a Wilcoxon rank test in R. Alpha index comparing among groups was computed by a Tukey's HSD test and a KruskalWallis H test in R. These analyses were conducted by Gene Denovo Co. (Guangzhou, China).
2.4 Total carbon (TC) and total organic carbon (TOC) analysisFirst, samples stored at -80℃ were freeze-dried, homogenized in an agate mortar, and kept in a desiccator. Elemental analyzer-mass spectrometer (Euro Vector, S.P.A.) was used to analyze the total carbon (TC) contents of samples. For the measurement of the total organic carbon (TOC) of sediment samples, about 150 mg sediment power was digested in 3 mL 10% HCl for 12 h at room temperature to remove inorganic carbon. The carbon of the residues was examined to obtain the TOC contents. The standard deviation of each measurement was determined by replicate analyses of the same sample.
3 RESULT AND DISCUSSION 3.1 TOC analysis of the sediment samplesOn sea-wide scales, the TOC contents are measured to reflect the distribution pattern of the accumulation and burial of the marine primary production (Seiter et al., 2004). Similar TOC contents were observed in the M30 and M94 samples at about 0.11%, and in M151 the TOC content decreased to 0.06%. Except for the M151 sample, the other two were consistent to those previously reported from the southern Mariana Trench (~6 000 m) (Luo et al., 2017), where the sedimentation rate was 0.02 cm per year. This rate is much higher than the averaged ones in deep ocean sediments (Luo et al., 2017). Although a lower TOC content was observed in the M151 sample, it is unclear whether a declined trend with increasing depths occurs in the southern region of the Mariana Trench. Alternatively, an accidental lower value may be observed, which was also found in another site of the southern part of the Mariana Trench where two evident spikes at 101 and 201 cm are present with TOC contents of 0.68% and 0.34%, respectively (Luo et al., 2017). However, the hadal shallowest sediment (down to 30 cm below the sea floor (cmbsf)) of the Mariana Trench contained higher TOC contents of 0.15%–0.23% (Yoshida et al., 2013). For the total inorganic carbon (TIC) contents, a similar pattern occurred, and no obvious difference was observed between M30 and M94 samples, which were higher than that of the M151 sample (Table 1). These data suggest different oceanographic backgrounds between the hadal region and the southern Mariana Trench.

Since different TOC contents were found in different sediment samples, we further examined the istribution of bacterial diversity in M30, M94 and M151 sediment samples by sequence analysis of the V3–V4 region of 16S rRNA gene. After sequencing and filtering potential erroneous sequences, over 80 000 tags were obtained from each sample. Subsequently, by removing the redundant tags, a total of about 397 245, 406 398, and 395 793 unique tags were obtained from M30, M94, and M151 samples, respectively (Table 2).

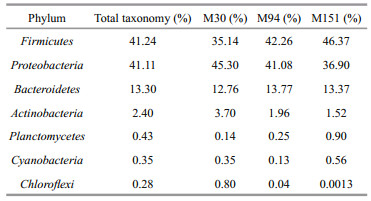
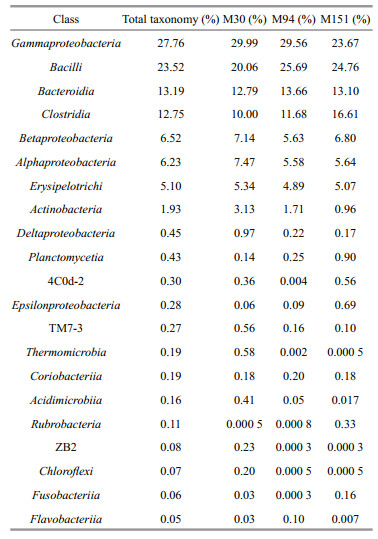
As shown in Fig. 2, the bacterial taxonomic richness, as examined by both numbers of OTUs and Chao1 estimates in sediment samples, is similar at the sites with different depths. Proteobacteria and Firmicutes dominated the prokaryotic 16S rRNA gene communities, which together combined for over 80% of the total community (Fig. 3a, Table 3). The OTUs of Proteobacteria decreased with depth, which could be resulted from the declined abundance of Gammaproteobacteria and Deltaproteobacteria. The OTU richness of Firmicutes increased at the deeper site due to higher abundance of Clostridia and Bacilli (Fig. 3b). Potentially phototrophic bacteria, such as Actinobacteria were also observed in all the samples and showed a decreasing trend with depth due to fewer OTUs of Actinobacteria and Acidimicrobia (Fig. 3b, Table 4). A similar trend was found in the group of Chloroflexi with a limited number of OTUs (Fig. 3a). The number of OTUs belonging to Planctomycetes was shown increased with depth. Bacteroidetes were present and exhibited similar richness in the three samples, indicating the photoheterotrophic metabolism occurs in the examined sediments. In addition, sizeable OTUs of Cyanobacteria were identified in all examined samples (Fig. 3a, Table 3).

|
| Fig.2 Comparison of OTU richness with depth Filled squares: numbers of OTUs; open squares: Chao1 richness values. |

|
| Fig.3 Bacterial community compositions of the sediments in the southern Mariana Trench at the levels of phylum (a) and class (b) |
Fifty-two classes were identified, and 21 were present in all samples counting for over 99% of the population. As shown in Table 4 and Fig. 3b, in M30, M94, and M151, the most abundant OTUs were Gammaproteobacteria and Bacilli, taking over 50% of the total tags. Bacteroidia and Clostridia combined for ~25% of the community. The bacterial population also contained Betaproteobacteria, Alphaproteobacteria, Erysipelotrichi, and Actinobacteria, and they together accounted for ~20%.
The bacterial community in the sediment of the southern Mariana Trench comprised a diversity of abundant groups. Most of OTUs including Gammaproteobacteria, Deltaproteobacteria, Alphaproteobacteria, Chloroflexi, Bacteroidetes, and Planctomycetia present in the hadal shallowest sediment (down to 30 cmbsf) of the Mariana Trench (Yoshida et al., 2013), were also observed in our samples. However, compared to the bacterial community in the hadal sediment, Gemmatimonadetes and Deferribacteres were not found, and instead, sizeable OTUs of Cyanobacteria and Actinobacteria occurred in our sediments. The different microbial compositions were caused probably by the different TOC contents between the hadal region and the southern region of the Mariana Trench. All of Gammaproteobacteria, Deltaproteobacteria, Alphaproteobacteria, Chloroflexi, Cyanobacteria, Bacteroidetes, Actinobacteria, and Planctomycetia identified in our samples are present in the water of the Challenger Deep (Nunoura et al., 2015; Tarn et al., 2016). Compared to reported bacterial OTUs in the Mariana Trench water (Nunoura et al., 2015; Tarn et al., 2016), we identified hardly the OTUs belonging to Nitrospirae and Marinimicrobia in our samples, a similar observation found in the hadal shallowest sediment of the Mariana Trench (Yoshida et al., 2013). This implies that different microbial compositions occur in the water and sediment of the Mariana Trench. Although Bacteroidetes from hadal water dominated the total tax tags in 6 000 mbs (meters below surface) only (Nunoura et al., 2015; Tarn et al., 2016), it accounted for a large number of OTUs in the sediments from 30, 94, and 151 cm in 4 000 mbs from the southern Mariana Trench and 30 cmbsf of the hadal region (Yoshida et al., 2013). Consistent with the observation of seawater samples containing a large number of the Gammaproteobacteria (Nunoura et al., 2015; Tarn et al., 2016), high abundance of this group was also identified in our sediment samples, indicating an effective heterotrophy. In addition, a limited number of OTUs belonging to sulfate reducer Desulfovibrio were observed in all samples from the sediments of the southern region of the Challenger Deep. Moreover, different from that large numbers of archaea were found in the water and the shallowest sediment of the Mariana Trench (Yoshida et al., 2013; Nunoura et al., 2015; Tarn et al., 2016), almost no OTUs belonging to archaea were observed in our sediment samples. Since we did not have more samples from the different depths of the location in this study or other sites of the regions of the Mariana Trench, and few datasets are available for the microbial diversity in the sediment of this region, it is unclear why archaea are missing in the sediments from the southern Mariana Trench.
4 CONCLUSIONThe bacterial communities in the abyssal sedimentary environments in the southern region of the Mariana Trench are quite complicated. In the samples of three depths of 30, 90, and 151 cm beneath the see floor, Proteobacteria and Firmicutes dominated the total taxon tags, followed by Bacteroidetes, Actinobacteria, Planctomycetes, Cyanobacteria, and Chloroflexi. These groups together combined up to over 99% of the total community. However, since few datasets about the bacterial communities in the sedimentary environments of the regions of the Mariana Trench are available, additional studies and more databases are required to reveal the composition of the microbes at the great depth.
5 DATA AVAILABILITY STATEMENTThe original sequence data that support the findings of this study have been deposited in the NCBI Sequence Read Archive (SRA) database with the primary accession number PRJNA449582.
6 ACKNOWLEDGMENTWe thank Prof. TIAN Jiwei and Prof. LIU Yanguang for providing the sediment samples.
DeSantis T Z, Hugenholtz P, Larsen N, Rojas M, Brodie E L, Keller K, Huber T, Dalevi D, Hu P, Andersen G L. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology, 72(7): 5069-5072.
DOI:10.1128/AEM.03006-05 |
Edgar R C. 2013. UPARSE:highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10): 996-998.
DOI:10.1038/nmeth.2604 |
Glud R N, Wenzhöfer F, Middelboe M, Oguri K, Turnewitsch R, Canfield D E, Kitazato H. 2013. High rates of microbial carbon turnover in sediments in the deepest oceanic trench on Earth. Nature Geoscience, 6(4): 284-288.
DOI:10.1038/ngeo1773 |
Kato C, Li L, Nogi Y, Nakamura Y, Tamaoka J, Horikoshi K. 1998. Extremely barophilic bacteria isolated from the Mariana Trench, Challenger Deep, at a depth of 11, 000 meters. Applied and Environmental Microbiology, 64(4): 1510-1513.
|
León-Zayas R, Peoples L, Biddle J F, Podell S, Novotny M, Cameron J, Lasken R S, Bartlett D H. 2017. The metabolic potential of the single cell genomes obtained from the Challenger Deep, Mariana Trench within the candidate superphylum Parcubacteria (OD1). Environmental Microbiology, 19(7): 2769-2784.
DOI:10.1111/emi.2017.19.issue-7 |
Luo M, Gieskes J, Chen L Y, Shi X F, Chen D F. 2017. Provenances, distribution, and accumulation of organic matter in the southern Mariana Trench rim and slope:Implication for carbon cycle and burial in hadal trenches. Marine Geology, 386: 98-106.
DOI:10.1016/j.margeo.2017.02.012 |
Nakanishi M, Hashimoto J. 2011. A precise bathymetric map of the world's deepest seafloor, Challenger Deep in the Mariana Trench. Marine Geophysical Research, 32(4): 455-463.
DOI:10.1007/s11001-011-9134-0 |
Nogi Y, Kato C. 1999. Taxonomic studies of extremely barophilic bacteria isolated from the Mariana Trench and description of Moritella yayanosii sp.nov., a new barophilic bacterial isolate.. Extremophiles, 3(1): 71-77.
DOI:10.1007/s007920050101 |
Nunoura T, Takaki Y, Hirai M, Shimamura S, Makabe A, Koide O, Kikuchi T, Miyazaki J, Koba K, Yoshida N, Sunamura M, Takai K. 2015. Hadal biosphere:insight into the microbial ecosystem in the deepest ocean on Earth. Proceedings of the National Academy of Sciences of the United States of America, 112(11): E1230-E1236.
DOI:10.1073/pnas.1421816112 |
Pathom-aree W, Stach J E M, Ward A C, Horikoshi K, Bull A T, Goodfellow M. 2006. Diversity of actinomycetes isolated from Challenger Deep sediment (10, 898 m) from the Mariana Trench. Extremophiles, 10(3): 181-189.
DOI:10.1007/s00792-005-0482-z |
Schloss P D, Westcott S L, Ryabin T, Hall J R, Hartmann M, Hollister E B, Lesniewski R A, Oakley B B, Parks D H, Robinson C J, Sahl J W, Stres B, Thallinger G G, van Horn D J, Weber C F. 2009. Introducing mothur:opensource, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23): 7537-7541.
DOI:10.1128/AEM.01541-09 |
Seiter K, Hensen C, Schröter E, Zabel M. 2004. Organic carbon content in surface sediments-defining regional provinces. Deep Sea Research Part I:Oceanographic Research Papers, 51(12): 2001-2026.
DOI:10.1016/j.dsr.2004.06.014 |
Takami H, Inoue A, Fuji F, Horikoshi K. 1997. Microbial flora in the deepest sea mud of the Mariana Trench. FEMS Microbiology Letters, 152(2): 279-285.
DOI:10.1111/j.1574-6968.1997.tb10440.x |
Tarn J, Peoples L M, Hardy K, Cameron J, Bartlett D H. 2016. Identification of free-living and particle-associated microbial communities present in hadal regions of the Mariana Trench. Frontiers in Microbiology, 7: 665.
DOI:10.3389/fmicb.2016.00665 |
Wang Q, Garrity G M, Tiedje J M, Cole J R. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16): 5261-5267.
DOI:10.1128/AEM.00062-07 |
Yoshida M, Takaki Y, Eitoku M, Nunoura T, Takai K. 2013. Metagenomic analysis of viral communities in (hado)pelagic sediments. PLoS One, 8(2): e57271.
DOI:10.1371/journal.pone.0057271 |