 2019, Vol. 37
2019, Vol. 37Institute of Oceanology, Chinese Academy of Sciences
Article Information
- GUO Li, LIANG Sijie, ZHANG Zhongyi, LIU Hang, WANG Songwen, YANG Guanpin
- Domestication of marine microalga Nannochloropsis oceanica to freshwater medium and the physiological responses
- Journal of Oceanology and Limnology, 37(4): 1353-1362
- http://dx.doi.org/10.1007/s00343-019-8202-1
Article History
- Received Aug. 6, 2018
- accepted in principle Oct. 5, 2018
- accepted for publication Nov. 3, 2018




2 Key Laboratory of Marine Genetics and Breeding of Ministry of Education, Ocean University of China, Qingdao 266003, China;
3 Institutes of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, China;
4 College of Agriculture and Resources and Environment, Tianjin Agricultural University, Tianjin 300384, China
Salinity, one of the major environmental factors, is a selection pressure against the evolution of organisms from saltwater to freshwater habitats and vice versa (Marschner, 1995; Sudhir and Murthy, 2004). To make it easy to describe what we have done in this study, we defi ne here "acclimation" as a temporary response of an organism to salinity change and "adaptation" as its permanent inhabitation in salinity changed environments. We also refer to the artifi cial evolutionary process adapting an organism to salinitychanged environments to "domestication" which is widely adopted by crop breeders. Domestication is actually a process of identifying and isolating (selecting) the most desirable and genetically unique variant which may either exist in the original population or appear newly during domestication. Salinity change subjects organisms to an osmotic stress where they must either adjust their physiological processes rapidly and temporarily (acclimation) or reprogram their gene expression pattern genetically and permanently (adaptation) or both.
Salt tolerance of crops is always one of the major traits that breeders try to improve. Similarly, domestication of microalgae to salinity changed cultivating conditions is highly appreciated by microalgal cultivators because the microalgae cultivated in salinity changed environments may change their performances and alleviate the damage of harmful organisms. To our knowledge (personnel communications), the microalgal species subjecting salinity domestication include at least Spirulina platensis , Tribonema sp. and Tetraselmis sp. among others in China at present. Domestication to different cultivating conditions (e.g. organic matters as carbon sources and high concentrations of CO2) has been evolving as a wide practice in microalgal cultivation.
There must be mechanisms for organisms to cope with the change of environmental salinity. These mechanisms have been correlated with many observations including structural characteristics, metabolic pathways and genetic modifi cations among others. An osmoregulation process exists in many aquatic animals like L . vannamei , where osmotic stress leads to cell shrinkage and swelling, aff ecting cell volume and then cytoskeletal organization (Pedersen et al., 2001). The cytoplasmic osmotic strength may interact with the cytoskeleton, maintaining cellular osmotic balance (Liu et al., 2013). The osmoregulation involves a number of physiological pathways including cross membrane transportation of ions and energy metabolism (Chen et al., 2015). Many phytoplankton may be able to adapt rapidly to abiotic stresses where they reproduce in either a rapidly changing climate or occasionally physical events. Their responses to salt stress include, for example, reduction in photosynthesis, upregulation of glycerophospholipid signaling and transcription and translation machinery (Perrineau et al., 2014), and proline may provide the resistance to salt stress in these species (Mastrobuoni et al., 2012). Reprograming gene expression pattern has been observed in the fast evolutionary adaptation in coccolithophore Emiliania huxleyi to ocean acidifi cation (Lohbeck et al., 2014). The permanent change of DNA sequence and genomic structure (Sunday et al., 2014) and epigenetic shifting (Crevillén et al., 2014) is the potential ways of regulating and reprograming gene expression pattern. The salt tolerance of plants varies between very saltsensitive glycophytes and highly salt tolerant halophytes, and the mechanisms underlining salt tolerance are diverse among species and variable in degree (Ji et al., 2013; Rozema and Schat, 2013).
Species in genus Nannochloropsis are widely used to study the accumulation of lipids. They are cultivated also as animal feeds and sources of feed or food additives like eicosapentaenoic acid. Of the six species in this genus, fi ve inhabit the seawater and one inhabits the freshwater (Fawley and Fawley, 2007). Marine Nannochloropsis species have been found to be able to maintain a very slow growth in freshwater (Huang and Hu, 2013). Nannochloropsis species may acclimate to freshwater fast by changing their gene expression pattern temporarily if fewer generations of cell division are allowed and adapt to freshwater by reprograming their gene expression pattern permanently if more generations of cell division are allowed. In this study, we first domesticated N . oceanica , a marine microalga, to freshwater medium, and isolated a set of strains that adapted to freshwater medium. We then documented the transcriptomes of the domesticated strain cultivated in freshwater medium and the original species exposed to freshwater medium to reveal the metabolic pathways aff ected by reprograming gene expression pattern. In the end, we identifi ed the differentially expressed genes (DEGs) between the strain adapted to and the original species exposed to the freshwater medium, and tried to understand the physiological changes correlated with the adaptation of N . oceanica to freshwater medium. This study was carried out in order to validate the feasibility of microalgal domestication in practice and verify the physiological modifi cation during microalgal domestication. Here in this paper, we present our fi ndings.
2 MATERIAL AND METHOD 2.1 Domestication of N . oceanica and microalgal cultivationMarine microalga N . oceanica LAMB0001 (wild type, WT) was provided by Key Laboratory of Mariculture of Chinese Ministry of Education, Ocean University of China. This species holds a monoploid nucleus and reproduces asexually (Galloway, 1990; Kilian et al., 2011; Pan et al., 2011). Its genome has been repeatedly sequenced (Vieler et al., 2012; Liang et al., 2013). The species in genus Nannochloropsis are mainly used as the feed of fish larvae and rotifers and the food additive for human nutrition. They have evolved gradually as the models of both industrial application and biological researches (Weeks, 2011; Gee and Niyogi, 2017).
Nannochloropsis oceanica LAMB0001 was cultivated for about 730 generations (~two days each generation) in media with their salinity decreased and the component changed gradually by increasing the replaced volume of f/2 medium (pH 7.8, salinity 30) (Guillard and Ryther, 1962; Guillard, 1975) with BG11 medium (Stanier et al., 1971), which were autoclaved at 121℃ for 30 min. During the process of domestication lasting about 4 years, one-half volume of culture was replaced by fresh BG11 each month, and the ratio of f/2 medium decreased exponentially. The microalga eventually adapted to BG11 and was maintained in BG11, a medium appropriate for and widely used to the cultivation of freshwater cyanobacteria, i.e., the microalga completely changed its habitat from f/2 prepared with seawater (collected far Qingdao shore, fi ltrated ahead of use) to BG11 prepared with freshwater (double distilled water). A group of algal colonies (stains hereafter) was isolated, and one of these strains (FA1) was used in this study.
WT was cultivated in f/2 and FA1 was cultivated in BG11 to the stationary growth phase, respectively, with their cells harvested through centrifugation at 4 000×g for 10 min and resuspended in f/2 and BG11. The growth curves were drawn after WT was inoculated in f/2 (WT) and BG11 (WT exposing to freshwater, WT-F), respectively, and FA1 was inoculated in f/2 (FA1 exposing to seawater, FA1-M) and BG11 (FA1), respectively. The inoculants were cultivated at 25℃ and under an irradiation of 70 μmol/ (m2·s) following a rhythm of 12 h dark and 12 h light. The OD750 was read every two days and used to draw the growth curve. In order to extract the total RNA, WT and FA1 were both inoculated into the BG11 medium, respectively, and cultivated under the same condition as that for drawing the growth curve for three days when the microalga was at its exponential growth stage. The microalgal cells were harvested through centrifugation at 4 000×g for 10 min, washed with BG11 and frozen immediately in liquid nitrogen and used to total RNA extraction.
2.2 Assigning the freshwater adapted strains to N . oceanicaNannochloropsis oceanica LAMB0001 and 3 freshwater-adapted strains, N . oceanica FA1, N . oceanica FA2, and N . oceanica FA3, cultivated in f/2 and BG11 media, respectively, were harvested at their stationary growth stage through centrifugation with their DNA extracted with E.Z.N.A.® Plant DNA Kit (Omega Bio-Tek, USA) following manufacturer's instructions. The PCR was carried out in a volume of 50 μL containing 1X LA PCR Buff er II (Mg2+ Plus), 0.4 mmol/L dNTP (each), 50 ng template DNA, 400 nmol/L 18S rRNA gene amplifi cation primers (each direction) and 2.5 U TaKaRa LA Taq DNA polymerase. The amplifi cation primers were newly designed, which were 5′-CTCTGAATCTGCGAATG-3′ and 5′-TTTAGATAACTTCTCACGCT-3′. The reaction was thermocycled by predenaturing at 98℃ for 1 min followed by 30 cycles of denaturing at 98℃ for 10 s, annealing at 57℃ for 10 s and extending at 72℃ for 2 min and an extra extension at 72℃ for 4 min. The PCR product was checked for the length and integrity through electrophoresis in 0.8% agarose gel, and sequenced commercially on an ABI3730x1 DNA analyzer with traditional chain termination method (Sanger method) with PCR amplifi cation primers forwardly and reversely and primers 5′-CCAGCTCCAATAGCGTAT-3′ and 5′-GGGAGTATGGTCGCAAGG- 3′ forwardly designed in the middle regions. The overlapping reads were direction transformed and assembled into complete 18S ribosomal RNA gene sequences with the primer corresponding regions and the low quality parts at both ends in the assembled trimmed. The 18S rRNA gene sequences of the six known species in genus Nannochloropsis and outgroup species were retrieved from GenBank, which were then aligned with the newly obtained in this study with ClustalX 2.1 (Posada, 2008). After trimming the regions corresponding to the missing parts at both ends of the newly obtained sequences in this study, the remained were used to screen the most appropriate phylogenetic tree calculation model with jModelTests (Larkin et al., 2007) and construct the phylogenetic tree with MrBayes 3.2 (Ronquist et al., 2012).
2.3 RNA extraction, library construction, and sequencingThe total RNA was extracted using Trizol Reagent (Invitrogen, USA) following the manufacturer's instructions. The total RNA was assessed for degradation through electrophoresis in 1% agarose gel. The concentration of RNA was measured with a Qubit2.0 Fluorometer. The purity and integrity of the RNA were determined with an Agilent 2100 Bioanalyzer. About three micrograms of RNA with RNA integrity (RIN) of ~8 was used for library construction, from which poly (A) tailed mRNA was isolated using the oligo(dT) magnetic beads in Oligotex mRNA Kits (Qiagen), fragmented and used as the template for the first-strand cDNA synthesis 1356 J. OCEANOL. LIMNOL., 37(4), 2019 Vol. 37 with reverse transcriptase and random hexamers. The second-strand cDNA was synthesized with RNase H and DNA polymerase Ⅰ. Following end polishing, each ds cDNA fragments was added an 'A' base at both ends and ligated with adapters. The modifi ed ds cDNA fragments were gel purifi ed and PCR amplifi ed, yielding a cDNA sequencing library which was quantifi ed with an Agilent 2100 Bioanalyzer.
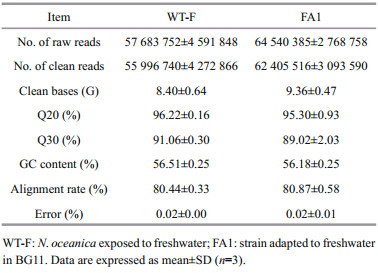
Transcriptome sequencing was carried out on an Illumina MiSeqTM platform (Novogene Bioinformatics Technology Co. Ltd.). The raw reads in FASTA format were processed through in-house perl scripts, yielding clean reads. During the processing, GC content and quality parameters including Q20 and Q30 were calculated. All the downstream analyses were based on the clean reads. The genome of N . oceanica (Vieler et al., 2012) was used as the reference with an index built using Bowtie v0.12.8 (Alverson, 2007). The clean reads were aligned onto the reference using Hisat2 (Kim et al., 2015).
2.4 Determination of differential transcriptsThe salinity was reduced from f/2 medium to BG11 medium. Simultaneously, the components of f/2 were changed gradually to those of BG11. In order to fi nd the genes with their expression patterns aff ected by salinity alone, the transcriptomes subjected to comparison were limited to the transcriptome of N . oceanica exposed to BG11 and that of N . oceanica adapted to BG11. This comparison made the infl uence of medium components the same and the infl uence of salinity alone.
Prior to the determination of differential transcripts, the reads were adjusted for each sequenced library with edgeR program package (3.22.3) through one scaling normalized factor. HTSeq v0.5.3 was used to count the reads mapped to each gene. The abundance of each transcript was normalized into TPM, a parameter indicating the abundance of a transcript (Wagner et al., 2012). Diff erential transcripts between WT-F and FA1 were determined with DESeq R package (1.32.0) (Anders and Huber, 2010). The adjusted P -value of 0.05 and log2|Foldchange| > 1.00 were set as the thresholds for differential transcripts. The P -values were adjusted to account for multiple testing by using the false discovery rate (FDR) (Benjamini and Yekutieli, 2001).
2.5 Pathway enrichments of differential transcriptsThe differential transcripts were enriched into Gene Ontology (GO) terms (function groups) with GOseq R Package (Young et al., 2010). GO terms with FRD of < 0.05 were considered signifi cant. These differential transcripts were also enriched into KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (http://www.genome.jp/kegg/) with KOBAS software (Mao et al., 2005).
3 RESULT 3.1 Microalgal domesticationBy cultivating marine N . oceanica LAMB0001 for about 730 generations (~ two days each generation) in media with salinity decreased and components changed gradually by increasing continuously the replaced volume of f/2 medium with the BG11 medium. The microalga eventually adapted to BG11, a medium appropriate for and widely used to cultivating cyanobacteria, i.e., from f/2 prepared with seawater to BG11 prepared with freshwater. A group of microalgal lines was isolated, and one of these lines (FA1) was used in this study.
The species in genus Nannochloropsis are very small in cell size and simple in morphological characteristics. The 18S ribosomal RNA gene sequence has been introduced into their classifi cation as an auxiliary marker. The three lines adapted to BG11 were most closely related to N . oceanica as their 18S ribosomal RNA gene sequences merged into a clade containing known N . oceanica (Fig. 1). Accordingly, three fresh water adapted lines of N . oceanica LAMB0001, FA1 through FA3, were assigned to species N . oceanica .

|
| Fig.1 Assigning three freshwater-adapted strains to N. oceanica according to their 18S ribosomal RNA gene sequences a. the variation among the 18S ribosomal RNA genes of the six species in genus Nannochloropsis and three freshwater-adapted stains obtained in this study; b. the phylogenetic tree constructed according to the 18S ribosomal RNA gene sequences of species in genus Nannochloropsis and three freshwater-adapted strains. |
WT grew normally in f/2 medium; however, its growth was extremely slow in BG11 medium (Fig. 2). FA1 grew faster in BG11 than WT did in f/2 medium, and reached a signifi cantly higher cell density at the end of cultivation. When FA1 was cultivated in f/2, it recovered a faster growth than WT in f/2 after a brief adjustment, and performed similarly to FA1 did in BG11. These fi ndings indicated that domestication of N . oceanica to freshwater medium improved its growth performance at least.

|
| Fig.2 The growth curves of wild type N . oceanica LAMB 0001 in f/2 (WT), WT exposed to BG11 (WT-F), freshwater adapted strain in BG11 (FA1) and FA 1 inoculated back to f/2 prepared with seawater (FA1-S) OD750 is recorded every two days, which is expressed as mean±SD (n =3). |
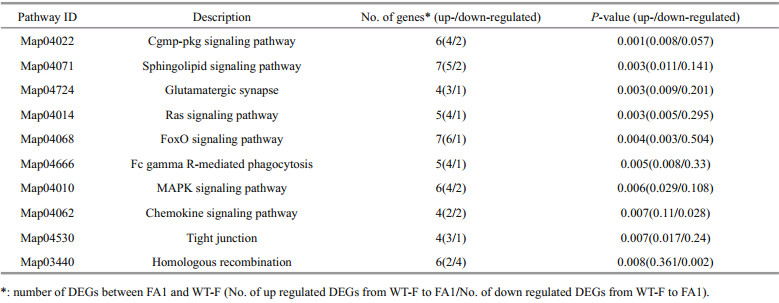
After discarding low quality reads, the clean ones (Table 1) were aligned (mapped) to the gene models of the reference genome. A total of 11 877 genes in the reference genome (99.17%) were aligned with > one read of either FA1 or WT-F. In total, 1 239 (10.43%) genes expressed differentially between FA1 and WT-F (Fig. 3). Of the genes, 393 were up-regulated expressed and 846 down-regulated expressed between FA1 and WT-F. In order to understand the function of differently expressed genes, they were enriched in GO term and KEGG pathway according to genome annotation information. In GO enrichment, the 118 DEGs were signifi cantly enriched in 12 GO terms including microtubule protein phosphorylation and membrane transport (GO: 0005887, GO: 0016020, GO: 0055085), phosphate ion transport (GO: 0022891, GO: 0005315, GO: 0015293, GO: 0006817), microtubule (GO: 0003777, GO: 0007018), protein phosphorylation (GO: 0006468), cell adhesion (GO: 0007155), chemotaxis to cAMP (GO: 0043327) (Table 2, Supplementary Table 1). Twenty-six DEGs were signifi cantly enriched in 10 KEGG pathway (Table 3, Supplementary Table 2), of which, 12 DEGs were also found in GO enrichment and most of them were related to protein phosphorylation. These expression patterns of DEGs are shown in the heatmap of genes different function (Fig. 3).

|
| Fig.3 The heatmap of genes differentially expressed between FA and WT-F a. the heatmap of all DEGs; b. the heatmaps of DEGs with different functions. |

|
When wild type N . oceanica is exposed to freshwater medium (WT-F), it will adjust its physiological metabolism rapidly and temporarily to respond the salinity changed environment while in BG11 medium, the freshwater medium adapted strain (FA1) physiologically performs in a way different from that of N . oceanica exposed to the BG11 medium. We assumed that the expression patterns of the genes differentially expressed between FA1 and WT-F have been reprogrammed, and the metabolic processes they correlate with have been genetically modifi ed. A GO term or a KEGG pathway came into our sights if it was enriched signifi cantly by DEGs between FA1 and WT-F. A GO term or a KEGG pathway enriched by DEGs between FA1 and WT-F was believed to be up- or down-regulated if it was enriched simultaneously by up- or down-regulated DEGs between FA1 and WT-F.
On this assumption, the up-regulated GO terms (corrected P < 0.05) covered cell adhesion, membrane, and membrane integrity and ion and substrate transportations while the down-regulated GO terms correlated with microtubule-based cell movement. Except for cell adhesion, the regulation of these terms indicated that the microalga got adapted to the freshwater medium by strengthening its ion and substrate transportations and simultaneously slowing down its movement to some extent. Only two terms (GO: 0006468 and GO: 0043327) were enriched with DEGs between FA1 and WT-F, but not raised according to the enrichment of up-regulated DEGs or lowered according to the enrichment of downregulated DEGs. Protein phosphorylation is widely involved in the modifi cation of protein functions. The enrichment of this term indicates that the microalga adapted to freshwater medium harmonized the relationship among its proteins and further its metabolism and performance. Why the microalga signifi cantly modifi ed its chemotaxis to cAMP was not very clear. Chemotaxis orients cells (or organisms) to respond to chemical gradients including, for example, food sources and invading cells. Extracellular cAMP attracts starving cells to aggregate to one another, initiating the developmental phase of the life cycle, which may cope with up-regulated cell adhesion and down-regulated microtubule motor activity and microtubule-based movement. Intracellular cAMP activates the catalytic activity of the cAMP-dependent protein kinase, triggering the expression of development-specifi c genes. The microalga may modify its chemotaxis to cAMP in order to adjust the growth and division of its cells in a freshwater medium; however, this understanding needs more supports from further studies
As listed in Table 3, of the enriched KEGG pathways (P < 0.01) by DEGs between FA1 and WT-F, fi ve signaling pathways were found to be up-regulated and one was found to be down-regulated. These pathways may have netted the signals, gene expression, cellular metabolism, and cell performance, indicating that N . oceanica fulfi lled its adaptation to freshwater medium mainly by changing its signaling net. For example, the MAPK pathway is a chain of proteins that link the receptors on the cell surface with the DNA in the cell nucleus, correlating with gene expression and cell division; and chemokines mediate a wide range of biological activities like chemotaxis and superoxide radical production through interactions with transmembrane receptors.
The up-regulated pathways included also tight junction (04530), glutamatergic synapse (04724) and Fc gamma R-mediated phagocytosis (04666). Tight junctions may serve purposes such as material transport and maintenance of osmotic balance. Glutamatergic synapse correlates with the synaptic transmission. Phagocytosis is essential for the uptake and destruction of infectious pathogens. Unfortunately, how these three animal-specifi c pathways function in the microalga and why they were up-regulated are not clear.
4 DISCUSSIONIn this study, a set of freshwater medium BG11 adapted lines of the marine microalga N . oceanica were obtained through domestication and verifi ed thorough the phylogenetic analysis of the 18S ribosomal RNA gene. The line FA1 grew faster in the BG11 medium than wild type N . oceanica grew in the f/2 medium. It may have reprogrammed the expression patterns of a set of genes that correlate with cell adhesion, membrane and membrane integrity, material transportation and cell movement and cellular signaling network. Our fi ndings indicated that the domestication of marine microalgae to freshwater habitat is feasible, which may aid in improving the microalgal cultivation performance. The most obvious characteristics of N . oceanica are its monoploidy and asexual reproduction mode (Pan et al., 2011). These characteristics make this species desirable for domestication.
In order to transfer truly the genetic information under changed conditions, evolution has off ered cells a highly organized and coordinated ability to deal with the DNA damage. Cells sense the problems in DNA and then activate DNA repairing. Domestication certainly makes organisms respond to artifi cially set stresses continuously. This will force cells to change their genome structure. Therefore, domestication should not be understood as a selection process alone, but also a mutation causing and favorite variant fi xing process. On this understanding, the range of genetic variation will be widened through mutation, and the effi ciency of domestication is increasable if a mutation step is integrated into this process.
Epigenetic shifting of gene expression may also have taken place during domestication of N . oceanica to freshwater. The expression of histone methyltransferase SETD3 gene was up-regulated (log2 Foldchange=1.37, padj=2.01E-07) in FA1 compared with WT-F. In eukaryotic cells, the genome is tightly condensed into chromatin composing of DNA and histone proteins (Kouzarides, 2007); such confi guration may be overcome by histone methyltransferases (Trievel et al., 2002). Histone methylation is important for the epigenetic modifi cation of chromatin; such modifi cation is associated with diverse cellular functions including, for example, gene expression, genomic stability, stem cell maturation, cell lineage development, genetic imprinting, DNA methylation and mitosis among others (Sawan and Herceg, 2010). Either temporally or permanently, change of chromatin confi guration should aff ect the expression of genes. The addition of methyl groups to a DNA changes its activity without changing its sequence. DNA methylation will repress the transcription and the expression pattern of a gene if the methylation is located in the promoter region. DNA methylation is associated with a number of cellular processes including, for example, genomic imprinting, chromosomal inactivation, and repression of transposable elements. Actually, chromatin confi guration, DNA methylation and their associations with the gene expression pattern have evolved as studying hot points recently. In this study, we found that the expression of histone methyltransferase encoding gene was extremely up-regulated, providing us a clue that chromatin confi guration may function during the domestication of N . oceanica to freshwater medium. Both DNA confi guration change and DNA sequence methylation will lead to genetic change during domestication. Unfortunately, more studies are appreciated for identifying and distinguishing these changes in DNA, the carrier of genetic information.
5 CONCLUSIONA number of BG11 medium adapted strains of marine N . oceanica were obtained through domestication and verifi ed phylogenetically based on 18S ribosomal RNA gene. It was found that BG11 adapted strain FA1 grew faster in the BG11 medium than wild type N . oceanica grew in the f/2 medium. FA1 may have reprogrammed the expression patterns of a set of genes that correlated with cell adhesion, membrane and membrane integrity, material transportation, cell movement, and cellular signaling network. It is feasible to domesticate marine microalgae to freshwater habitat, which may aid in improving their cultivation performance.
6 DATA AVAILABILITY STATEMENTThe data that support the fi ndings of this study are available from the corresponding author on request.
Electronic supplementary material
Supplementary material (Supplementary Tables 1–2) is available in the online version of this article at https://doi.org/10.1007/s00343-019-8202-1.
Alverson A J. 2007. Strong purifying selection in the silicon transporters of marine and freshwater diatoms. Limnology and Oceanography, 52(4): 1 420-1 429.
DOI:10.4319/lo.2007.52.4.1420 |
Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biology, 11: R106.
DOI:10.1186/gb-2010-11-10-r106 |
Benjamini Y, Yekutieli D. 2001. The control of the false discovery rate in multiple testing under dependency. The Annals of Statistics, 29(4): 1 165-1 188.
DOI:10.1214/aos/1013699998 |
Chen K, Li E C, Li T Y, Xu C, Wang X D, Lin H Z, Qin J G, Chen L Q. 2015. Transcriptome and molecular pathway analysis of the hepatopancreas in the Pacific white shrimp Litopenaeus vannamei under chronic low-salinity stress. PLoS One, 10(7): e0131503.
DOI:10.1371/journal.pone.0131503 |
Crevillén P, Yang H C, Cui X, Greeff C, Trick M, Qiu Q, Cao X F, Dean C. 2014. Epigenetic reprogramming that prevents transgenerational inheritance of the vernalized state. Nature, 515(7528): 587-590.
DOI:10.1038/nature13722 |
Fawley K P, Fawley M W. 2007. Observations on the diversity and ecology of freshwater Nannochloropsis(Eustigmatophyceae), with descriptions of new taxa. Protist, 158(3): 325-336.
DOI:10.1016/j.protis.2007.03.003 |
Galloway R E. 1990. Selective conditions and isolation of mutants in salt-tolerant, lipid-producing microalgae. Journal of Phycology, 26(4): 752-260.
DOI:10.1111/j.0022-3646.1990.00752.x |
Gee C W, Niyogi K K. 2017. The carbonic anhydrase CAH1is an essential component of the carbon-concentrating mechanism in Nannochloropsis oceanicav. Proceedings of the National Academy of Sciences of the United States of America, 114(17): 4 537-4 542.
DOI:10.1073/pnas.1700139114 |
Guillard R R L, Ryther J H. 1962. Studies of marine planktonic diatoms:I. Cyclotella nana Hustedt, and Detonula confervacea (Cleve) gran. Canadian Journal of Microbiology, 8(2): 229-239.
|
Guillard R R L. 1975. Culture of phytoplankton for feeding marine invertebrates. In:Smith W L, Chanley M H eds.Culture of Marine Invertebrate Animals. Plenum Press, New York. 29-60.
|
Huang W C, Hu H H. 2013. Study on the salinity tolerance and oil accumulation in Nannochloropsis. ActaHydrobiologica Sinica, 37(2): 383-387.
(in Chinese) |
Ji H T, Pardo J M, Batelli G, Van Oosten M J, Bressan R A, Li X. 2013. The Salt Overly Sensitive (SOS) pathway:established and emerging roles. Molecular Plant, 6(2): 275-286.
|
Kilian O, Benemann C S E, Niyogi K K, Vick B. 2011. Highefficiency homologous recombination in the oil-producing alga Nannochloropsis sp. Proceedings of the National Academy of Sciences of the United States of America, 108(52): 21 265-21 269.
DOI:10.1073/pnas.1105861108 |
Kim D, Langmead B, Salzberg S L. 2015. HISAT:a fast spliced aligner with low memory requirements. Nature Methods, 12(4): 357-360.
DOI:10.1038/nmeth.3317 |
Kouzarides T. 2007. Chromatin modifications and their function. Cell, 128(4): 693-705.
DOI:10.1016/j.cell.2007.02.005 |
Larkin M A, Blackshields G, Brown N P, Chenna R, McGettigan P A, McWilliam H, Valentin F, Wallace I M, Wilm A, Lopez R, Thompson J D, Gibson T J, Higgins D G. 2007. Clustal W and Clustal X version 2.0. Bioinformatics, 23(21): 2 947-2 948.
DOI:10.1093/bioinformatics/btm404 |
Liang C W, Cao S N, Zhang X W, Zhu B H, Su Z L, Xu D, Guang X Y, Ye N H. 2013. De novo sequencing and global transcriptome analysis of Nannochloropsis sp.(Eustigmatophyceae) following nitrogen starvation. BioEnergy Research, 6(2): 494-505.
|
Liu S K, Wang X L, Sun F Y, Zhang J R, Feng J B, Liu H, Rajendran K V, Sun L Y, Zhang Y, Jiang Y L, Peatman E, Kaltenboeck L, Kucuktas H, Liu Z J. 2013. RNA-Seq reveals expression signatures of genes involved in oxygen transport, protein synthesis, folding, and degradation in response to heat stress in catfish. Physiological Genomics, 45(12): 462-476.
DOI:10.1152/physiolgenomics.00026.2013 |
Lohbeck K T, Riebesell U, Reusch T B H. 2014. Gene expression changes in the coccolithophore Emiliania huxleyi after 500 generations of selection to ocean acidification. Proceedings of the Royal Society B:Biological Sciences, 281(1786): 20140003.
DOI:10.1098/rspb.2014.0003 |
Mao X Z, Cai T, Olyarchuk J G, Wei L P. 2005. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics, 21(19): 3 787-3 793.
DOI:10.1093/bioinformatics/bti430 |
Marschner H. 1995. Mineral Nutrition of Higher Plants. Academic Press, London.
|
Mastrobuoni G, Irgang S, Pietzke M, Aßmus H E, Wenzel M, Schulze W X, Kempa S. 2012. Proteome dynamics and early salt stress response of the photosynthetic organism Chlamydomonas reinhardtii. BMC Genomics, 13: 215.
DOI:10.1186/1471-2164-13-215 |
Pan K H, Qin J J, Li S, Dai W K, Zhu B H, Jin Y C, Yu W G, Yang G P, Li D F. 2011. Nuclear monoploidy and asexual propagation of Nannochloropsis oceanica(Eustigmatophyceae) as revealed by its genome sequence. Journal of Phycology, 47(6): 1 425-1 432.
DOI:10.1111/jpy.2011.47.issue-6 |
Pedersen S F, Hoffmann E K, Mills J W. 2001. The cytoskeleton and cell volume regulation. Comparative Biochemistry and Physiology Part A:Molecular & Integrative Physiology, 130(3): 385-399.
|
Perrineau M M, Zelzion E, Gross J, Price D C, Boyd J, Bhattacharya D. 2014. Evolution of salt tolerance in a laboratory reared population of Chlamydomonas reinhardtii. Environmental Microbiology, 16(6): 1755-1766.
DOI:10.1111/emi.2014.16.issue-6 |
Posada D. 2008. jModelTest:phylogenetic model averaging. Molecular Biology and Evolution, 25(7): 1 253-1 256.
DOI:10.1093/molbev/msn083 |
Ronquist F, Teslenko M, van der Mark P, Ayres D L, Darling A, Höhna S, Larget B, Liu L, Suchard M A, Huelsenbeck J P. 2012. MrBayes 3.2:efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61(3): 539-542.
DOI:10.1093/sysbio/sys029 |
Rozema J, Schat H. 2013. Salt tolerance of halophytes, research questions reviewed in the perspective of saline agriculture. Environmental and Experimental Botany, 92: 83-95.
DOI:10.1016/j.envexpbot.2012.08.004 |
Sawan C, Herceg Z. 2010. Histone modifications and cancer. Advances in Genetics, 70: 57-85.
DOI:10.1016/B978-0-12-380866-0.60003-4 |
Stanier R Y, Kunisawa R, Mandel M, Cohen-Bazire G. 1971. Purification and properties of unicellular blue-green algae(order Chroococcales). Bacteriological Reviews, 35(2): 171-205.
|
Sudhir P, Murthy S D S. 2004. Effects of salt stress on basic processes of photosynthesis. Photosynthetica, 42(4): 481-486.
DOI:10.1007/S11099-005-0001-6 |
Sunday J M, Calosi P, Dupont S, Munday P L, Stillman J H, Reusch T B H. 2014. Evolution in an acidifying ocean. Trends in Ecology & Evolution, 29(2): 117-125.
|
Trievel R C, Beach B M, Dirk L M A, Houtz R L, Hurley J H. 2002. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell, 111(1): 91-103.
|
Vieler A, Wu G X, Tsai C H, Bullard B, Cornish A J, Harvey C, Reca I B, Thornburg C, Achawanantakun R, Buehl C J, Campbell M S, Cavalier D, Childs K L, Clark T J, Deshpande R, Erickson E, Armenia Ferguson A, Handee W, Kong Q, Li XB, Liu B S, Lundback S, Peng C, Roston R L, Sanjaya, Simpson J P, TerBush A, Warakanont J, Zäuner S, Farre E M, Hegg E L, Jiang N, Kuo M H, Lu Y, Niyogi K K, Ohlrogge J, Osteryoung K W, Shachar-Hill Y, Sears B B, Sun YN, Takahashi H, Yandell M, Shiu S H, Benning C. 2012. Genome, functional gene annotation, and nuclear transformation of the heterokont oleaginous alga Nannochloropsis oceanica CCMP1779. PLoS Genet, 8(11): e1003064.
DOI:10.1371/journal.pgen.1003064 |
Wagner G P, Kin K, Lynch V J. 2012. Measurement of mRNA abundance using RNA-seq data:RPKM measure is inconsistent among samples. Theory in Biosciences, 131(4): 281-285.
DOI:10.1007/s12064-012-0162-3 |
Weeks D P. 2011. Homologous recombination in Nannochloropsis:a powerful tool in an industrially relevant alga. Proceedings of the National Academy of Sciences of the United States of America, 108(52): 20 859-20 860.
DOI:10.1073/pnas.1118670109 |
Young M D, Wakefield M J, Smyth G K, Oshlack A. 2010. Gene ontology analysis for RNA-seq:Accounting for selection bias. Genome Biology, 11(2): R14.
|