 2020, Vol. 38
2020, Vol. 38Institute of Oceanology, Chinese Academy of Sciences
Article Information
- JIANG Xiaoli, JING Xiaohong, LÜ Yan, SHAN Changmin, LI Jiahua, YU Yan, WU Yuyong, SUN Yeying
- Genome-wide identification and prediction of long noncoding RNAs in half-smooth tongue sole Cynoglossus semilaevis
- Journal of Oceanology and Limnology, 38(1): 226-235
- http://dx.doi.org/10.1007/s00343-019-8287-6
Article History
- Received Oct. 17, 2018
- accepted in principle Jan. 29, 2019
- accepted for publication Mar. 21, 2019




2 College of Life Science, Yantai University, Yantai 264005, China
Half-smooth tongue sole (Cynoglossus semilaevis) is a commercial flatfish, ranking among the most valuable fishes in the coastal areas of China. Similar to other flatfish species, this animal possesses an asymmetric body shape with lateralization of the eyes to the same side during metamorphosis (Chen et al., 2014).
In contrast to other flatfish species, half-smooth tongue sole features some unique natural characteristics. In nature, the length and weight of mature females are 2–4 times of mature males, and the mature ovary reaches more than one thousand times the volume and weight of the testis (Sun et al., 2010; Li et al., 2018). This species employs a female heterogametic sex determination system (ZW female and ZZ male) (Chen et al., 2009, 2014; Shao et al., 2014). However, the molecular mechanism that regulates these special characteristics remains unknown.
Long noncoding RNAs (lncRNAs) are defined as noncoding RNAs longer than ~200 nt and lack protein-encoding capacity (Philippen et al., 2015; Santosh et al., 2015). To date, more than 10 000 lncRNAs have been identified in humans, and their number continually grows. However, information on fishes remains lacking. LncRNAs are classified into five subclasses based on their location and function, namely intergenic, intronic, sense overlapping, antisense, and bidirectional lncRNAs. In contrast to microRNAs, studies of lncRNAs are still in their infancy due to extremely complex and complicated gene regulation mechanisms (Jiang and Ning, 2015; Philippen et al., 2015; Santosh et al., 2015; Lorenzen and Thum, 2016). However, increasing evidence has shown that lncRNAs perform strong biological functions and may be involved in all biological and developmental processes (Sun and Kraus, 2015).
In this study, the lncRNA profile in adult halfsmooth tongue soles was investigated using highthroughput sequencing method. We also analyzed the expression patterns of some lncRNAs in different tissues by quantitative reverse transcriptionpolymerase chain reaction (qRT-PCR).
2 MATERIAL AND METHOD 2.1 Fish sampling and RNA isolationFour mature female and four mature male halfsmooth tongue soles were obtained from a nearby hatchery (Laizhou City, Shandong Province, China). The fishes were anesthetized with MS-222 before being sampled. The gonad, liver, heart, kidney, gill, muscle, and brain tissues were collected individually and frozen at -80℃ (Table 1). In all experiments, total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). All tissues from mature female and male fishes (n=4) were pooled together to generate total RNA for high-throughput sequencing experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee of Binzhou Medical University.

The transcriptome of lncRNAs and mRNAs was determined via next-generation deep sequencing using Illumina platforms. Normal RNA-seq library was constructed. According to the manufacturer's transcriptome sequencing protocol, first strand cDNA was synthesized using random hexamer primers and M-MuLV Reverse Transcriptase. DNA polymerase I and RNase H were used to synthesize the second strand cDNA. After adenylation of the 3ʹ ends of the DNA fragments, an adaptor with a hairpin loop structure was ligated to prepare for hybridization. To select cDNA fragments with the preferred length of 150–200 bp, the library fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, CA, USA). Finally, quality was assessed on an Agilent Bioanalyzer 2100 system. The library was sequenced at the Annoroad Genome (Beijing, China) on an Illumina Hiseq 2000 platform.
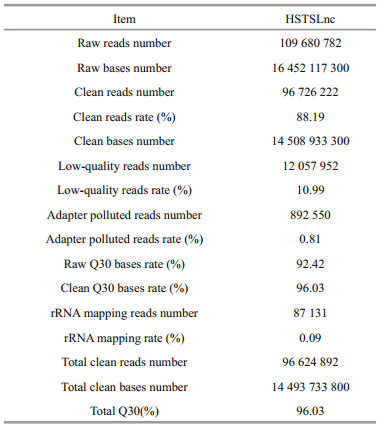
2.3 Quality control and sequencing data analysisThe sequencing data discussed in this publication have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE98425. To assess the quality of RNA-seq data, each base in the reads was assigned a quality score (Q) by Fast QC with a Phred-like algorithm. Clean data were obtained by removing reads containing adapters, reads containing over 10% poly-N, rRNA mapping reads and low-quality reads (> 50% of bases whose Q scores were < 20%) from the raw data. The clean data were mapped to the C. semilaevis reference genome (https://www.ncbi.nlm.nih.gov/genome/?term=Cynoglossus+semilaevis, GCA_000523025.1 Cse_v1.0) by TopHat2 (v2.0.13) with "-library-type fr-firststrand" as a parameter (Trapnell et al., 2012). The transcripts were assembled using Cufflinks (v2.2.0) according to the instructions provided (Trapnell et al., 2012).
2.4 LncRNA predictionTo identify lncRNA genes from the assembly results, the following criteria were considered. Firstly, transcripts shorter than 200 nt and single exons of transcripts were removed. Secondly, transcripts with an open reading frame that was longer than 300 nt were removed. Next, the transcripts with low expression levels (fragments per kilobase of transcript per million fragments mapped, FPKM < 1) and minimal read coverage threshold below 3 were discarded. Transcripts that overlapped with known protein-coding genes on the same strand were discarded. Then, Coding-Potential Assessment Tool (CPAT) (Wang et al., 2013), CodingNon-Coding Index (CNCI) (Sun et al., 2013) and Coding Potential Calculator (CPC) (Kong et al., 2007), were used to distinguish mRNA from lncRNAs. CPAT was used to assess the long noncoding transcripts by a logistic regression model trained by CPAT software based on the coding and noncoding transcripts of C. semilaevis from NCBI. The coding probability score from CPAT of C. semilaevis was optimized according to the instructions provided. Finally, the optimum cutoff for C. semilaevis gene annotation was determined as 0.356. Scores < 0.356 were retained as putative noncoding RNAs. CNCI software was downloaded from the web (https://github.com/www-bioinfo-org/CNCI) and used with the default parameters to predict noncoding RNAs. CPC (http://cpc.cbi.pku.edu.cn/) was used to predict the protein-coding potential for transcripts. Only transcripts with a CPC score ≤-1 were retained. The transcripts that remained were considered putative lncRNAs. The transcripts were submitted to identify known and novel lncRNAs by comparison against the NCBI-NR database.
2.5 qRT-qPCR of selected lncRNAsUsing specific primers designed for selected novel lncRNAs isolated from the library (Table 2), RT-PCR was performed to verify differential expression in seven tissues (gill, heart, brain, liver, muscles, kidneys and ovary) of female fish. The specific primers were designed by Primer Premier 5.0 software based on the sequencing results. Total RNAs from different tissues from female fishes (n=6) were reverse-transcribed to cDNA using SuperScript Ⅳ Reverse Transcriptase (ThermoFisher, USA). LncRNAs was performed on cDNAs using TaqManTM Fast Advanced Master Mix (ThermoFisher, USA). β-actin was used for internal control. The relative gene expression of selected lncRNAs was calculated by comparing the threshold cycle (Ct).

|
A total of 109 680 782 raw reads were produced from the cDNA libraries. After quality control, 96 726 222 clean reads were obtained. The proportion of reads with a Phred quality value higher than 30 among the clean reads totaled 96.03% (Table 3). Overall, 88.00% of the clean reads were aligned against the Cynoglossus semilaevis reference genome. Among these mapped reads, 80.7% of the reads were mapped to exon regions, 7.4% to intron regions, and 11.9% to intergenic regions (Fig. 1). According to their genome positions, these reads were widely distributed in all chromosomes of the half-smooth tongue sole (Fig. 2).

|
| Fig.1 Distribution of all mapped reads on half-smooth tongue sole genome |

|
| Fig.2 Genome-wide distribution of half-smooth tongue sole mapped reads Chromosomes are indicated in purple and in circular form as the outer track. In the next two tracks, abundances of mapped reads are indicated by orange and cyan columns. The orange track represents the distribution on the sense strand, and the cyan track represents distribution on the antisense strand. |
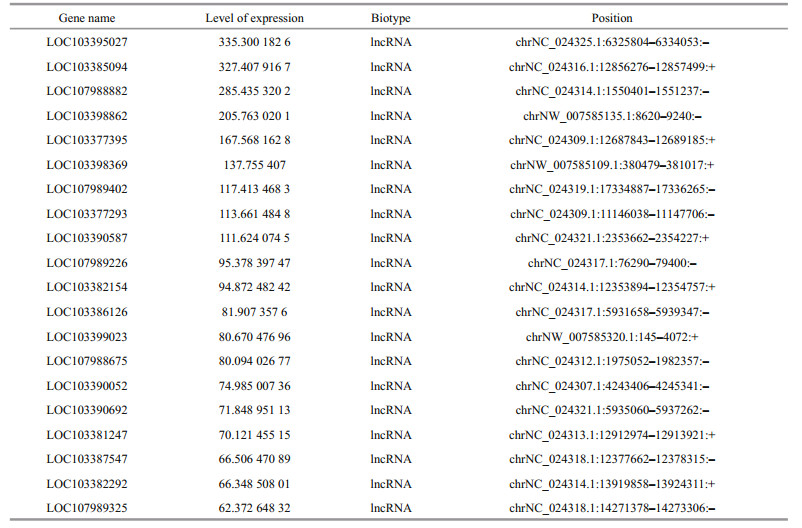
Transcripts were reconstructed using Cufflinks. To obtain a unique set of isoforms, all the reconstructed transcripts were assembled using Cuffcompare against the C. semilaevis reference genome, Cse_ v1.0. Potential lncRNAs were identified based on their sequences, amino acid peptide lengths, and protein-coding potential using CPAT, CNCI, and CPC methods. In this report, 1 694 known lncRNAs were identified from the sequencing library by BLAST in the NCBI-NR database. Table 4 lists the top 20 most abundant known lncRNAs. In addition to known lncRNAs, novel lncRNAs were predicted from the total assembled transcripts.
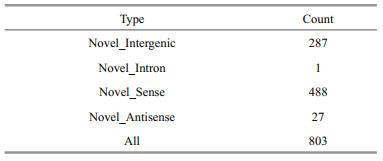
After removing low-abundance transcripts, known transcripts, CPAT-removed transcripts, CNCIremoved transcripts and CPC-removed transcripts, 1 412 potential lncRNA transcripts were obtained, and the results are shown in Table 5. Finally, 803 novel lncRNAs were identified; these novel lncRNAs included 287 novel intergenic, 1 novel intron, 488 novel senses, and 27 novel antisense types (Table 6). We analyzed the sequence length and exon number of novel lncRNAs. Results show that sequence length was mainly distributed in the range of 200 nt to 3 000 nt, and most lncRNAs contained 2–6 exons (Fig. 3). A total of 22 104 protein-coding transcripts were identified, and analysis of sequence length revealed that lncRNAs were shorter in sequence length and featured fewer exons than mRNAs (Fig. 4).


|
| Fig.3 Length and exons of novel lncRNAs a. distribution of length of novel lncRNAs. The majority of lncRNAs have less than 4 000 bp; b. distribution of the number of exons of novel lncRNAs. Most of the lncRNAs have 2-6 exons. |

|
| Fig.4 Structural comparison of novel lncRNAs and mRNAs a. distribution of the number of exons of novel lncRNAs and mRNAs. LncRNAs have a fewer number of exons; b. the sequence length of lncRNAs and mRNAs. On average, lncRNAs have shorter transcripts. |
To validate our deep sequencing results, six novel lncRNAs identified by deep sequencing analysis were selected. Their expression levels in different tissues of female fish were further determined by qRT-PCR. Table 2 lists the primers used in the study. Expression patterns of these lncRNAs showed dissimilarity in the tissues of female fish (Fig. 5). Lnc_673 and lnc_40 showed broad expressions and were both expressed in all tissue samples. Lnc_190, lnc_493, lnc_770 and lnc_150 showed characteristic tissue-specific expressions. Lnc_190 was strongly expressed in the brain and comparatively weaker in the liver of females. Lnc_493 was mainly expressed in brain and ovary compared with other female tissues. Lnc_770 and lnc_150 were mainly expressed in the ovary and slightly expressed in the liver and muscles of the female fish. These results imply that the two novel lncRNAs may be involved in the development of ovary in the fish.

|
| Fig.5 The expression level of six novel lncRNAs in seven tissues of female fish (n=6) All experiments were repeated at least three times. The data are shown as means±SD. |
In the past decades, significant genetic improvements have been achieved in half-smooth tongue soles. However, all aspects were focused on mRNAs and microRNA research (Liu et al., 2016a, b; Xu et al., 2016; Yan et al., 2016). Information on lncRNAs in half-smooth tongue soles remains lacking. The mechanisms and functions of lncRNAs have also remained unclear in marine fish. LncRNAs are noncoding RNAs longer than 200 nt; significant progress has been achieved in the study of these molecules in mice, humans and other model organisms in the last few years. Studies have shown that lncRNAs regulate metabolic tissue development and function, including muscle differentiation, microvascular dysfunction, colorectal cancer progression and vascular development (Gong et al., 2015; Viereck et al., 2015; Yan et al., 2015; Ma et al., 2016). Although numerous studies have indicated the importance of lncRNAs in gene regulation, little is known about their biological function in marine fishes. Our study is the first to screen for lncRNAs in half-smooth tongue sole (C. semilaevis) using largescale deep sequencing. After quality control, 96 726 222 clean reads were obtained, and 88.00% of the clean reads were successfully mapped to the C. semilaevis reference genome. As the first study of lncRNAs in C. semilaevis, we identified 803 novel lncRNAs. Six novel lncRNAs were selected for expression analysis by qRT-PCR. Some lncRNAs showed tissue-specific expression, indicating that lncRNAs may be involved in tissue development.
According to expression profile results by qRTPCR, the novel lncRNA lnc_190 was strongly expressed in the brain, lnc_493 was mainly expressed in the brain and ovary, whereas lnc_770 and lnc_150 were mainly expressed in the ovary. Numerous studies showed that lncRNA genes are expressed in tissuespecific patterns (Roberts et al., 2014). In studies of human cell lines, results show that 29% of lncRNAs were expressed specifically in a single-cell type, and only 10% were expressed in all cell types (Roberts et al., 2014). For example, in mice, the linc-Brn1b expression is primarily restricted to specific brain regions and predominantly restricted in the developing cerebral cortex. linc-Brn1b shows spatiotemporally regulated patterns of expression during cortical development (Sauvageau et al., 2013). These results support the attribution of potential function based on tissue-specific expression. The expression of lncRNAs is regulated both at the transcriptional and posttranscriptional levels. However, how tissue-specific expression is achieved is remains unclear, especially in fish. Transcription factors and miRNAs targeting each lncRNA might have some contribution to tissuespecific expression. LncRNA transcription could affect the expression of adjacent genes in cis, hybridize to the overlapping sense transcript, act as ceRNA, or function in more complex ways to regulate gene expression. In cotton (Gossypium hirsutum L.), expression patterns analysis showed that most intronic lncRNAs (75.95%) exhibit a consistent expression pattern with their adjacent protein-coding genes (Lu et al., 2016). C2 calcium-dependent domaincontaining protein 4C-like (C2cd4c) is an adjacent gene to lnc_190 on chromosome 6 in C. semilaevis. C2cd4c is overexpressed in the whole brain in other species (Yue et al., 2014). This implies lnc_190 and C2cd4c might have a certain regulation relationship. The hypothalamic-pituitary-gonadal axis plays an important role in reproduction regulation in fish (Shao et al., 2014). However, the regulatory mechanism remains unclear, especially in C. semilaevis. These novel lncRNAs showed tissue-specific expression patterns in the brain or ovary. Thus, lncRNAs may be involved in reproduction regulation in C. semilaevis. The detailed function and mechanisms require further studies.
Compared with those in mouse and humans, the function of lncRNAs in marine fish poses considerable research difficulties. However, with some newly emerging technologies, the function of lncRNAs in marine fish will be easily studied. Recently the technology of genome editing has been successfully used in C. semilaevis to study gene functions (Cui et al., 2017).
5 CONCLUSIONNon-coding RNAs comprise the majority of the transcriptome, whose important functions have been discovered in model organisms. To date, few lncRNAs have been characterized in marine fish, including half-smooth tongue sole. In this study, hundreds of lncRNAs were identified by highthroughput sequencing and bioinformatics-analyses. A total of 1 694 known lncRNAs and 803 novel lncRNAs were identified. Some lncRNAs showed differential tissue distribution. These results suggest that lncRNAs might be important regulatory factors in the development of the fish.
6 DATA AVAILABILITY STATEMENTThe raw reads of the present study were also uploaded to the Sequence Read Archive databases of NCBI with accession number SRR5494302 (NCBI BioProject accession number, PRJNA385074). This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession number GFXK00000000. The version described in this paper is the first version, GFXK01000000.
7 COMPETING INTERESTThe authors declare there are no competing interests.
Chen S L, Tian Y S, Yang J F, Shao C W, Ji X S, Zhai J M, Liao X L, Zhuang Z M, Su P Z, Xu J Y, Sha Z X, Wu P F, Wang N. 2009. Artificial gynogenesis and sex determination in half-smooth tongue sole (Cynoglossus semilaevis). Marine Biotechnology, 11(2): 243-251.
DOI:10.1007/s10126-008-9139-0 |
Chen S, Zhang G J, Shao C W, Huang Q F, Liu G, Zhang P, Song W T, An N, Chalopin D, Volff J N, Hong Y H, Li Q Y, Sha Z X, Zhou H L, Xie M S, Yu Q L, Liu Y, Xiang H, Wang N, Wu K, Yang C G, Zhou Q, Liao X L, Yang L F, Hu Q M, Zhang J L, Meng L, Jin L J, Tian Y S, Lian J M, Yang J F, Miao G D, Liu S S, Liang Z, Yan F, Li Y Z, Sun B, Zhang H, Zhang J, Zhu Y, Du M, Zhao Y W, Schartl M, Tang Q S, Wang J. 2014. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nature Genetics, 46(3): 253-260.
DOI:10.1038/ng.2890 |
Cui Z K, Liu Y, Wang W W, Wang Q, Zhang N, Lin F, Wang N, Shao C W, Dong Z D, Li Y Z, Yang Y M, Hu M Z, Li H L, Gao F T, Wei Z F, Meng L, Liu Y, Wei M, Zhu Y, Guo H, Cheng C H K, Schartl M, Chen S L. 2017. Genome editing reveals dmrt1 as an essential male sex-determining gene in Chinese tongue sole (Cynoglossus semilaevis). Scientific Reports, 7: 42 213.
DOI:10.1038/srep42213 |
Edgar R, Domrachev M, Lash A E. 2002. Gene expression omnibus:NCBI gene expression and hybridization array data repository. Nucleic Acids Research, 30(1): 207-210.
DOI:10.1093/nar/30.1.207 |
Gong C G, Li Z Z, Ramanujan K, Clay I, Zhang Y Y, LemireBrachat S, Glass D J. 2015. A long non-coding RNA, LncMyoD, regulates skeletal muscle differentiation by blocking IMP2-mediated mRNA translation. Development Cell, 34(2): 181-191.
DOI:10.1016/j.devcel.2015.05.009 |
Jiang X Y, Ning Q L. 2015. The emerging roles of long noncoding RNAs in common cardiovascular diseases. Hypertension Research, 38(6): 375-379.
DOI:10.1038/hr.2015.26 |
Kong L, Zhang Y, Ye Z Q, Liu X Q, Zhao S Q, Wei L P, Gao G. 2007. CPC:assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research, 35(S2): W345-W349.
|
Li J H, Lv Y, Liu R R, Yu Y, Shan C M, Bian W H, Jiang J, Zhang D L, Yang C, Sun Y Y. 2018. Identification and characterization of a conservative W chromosome-linked circRNA in half-smooth tongue sole (Cynoglossus semilaevis) reveal its female-biased expression in immune organs. Fish & Shellfish Immunology, 82: 531-535.
|
Liu J X, Zhang W, Du X X, Jiang J J, Wang C L, Wang X B, Zhang Q Q, He Y. 2016a. Molecular characterization and functional analysis of the GATA4 in tongue sole(Cynoglossus semilaevis). Comparative Biochemistry and Physiology Part B:Biochemistry and Molecular Biology, 193: 1-8.
DOI:10.1016/j.cbpb.2015.12.001 |
Liu J X, Zhang W, Sun Y, Wang Z G, Zhang Q Q, Wang X B. 2016b. Molecular characterization and expression profiles of GATA6 in tongue sole (Cynoglossus semilaevis). Comparative Biochemistry and Physiology Part B:Biochemistry and Molecular Biology, 198: 19-26.
DOI:10.1016/j.cbpb.2016.03.006 |
Lorenzen J M, Thum T. 2016. Long noncoding RNAs in kidney and cardiovascular diseases. Nature Reviews Nephrology, 12(6): 360-373.
DOI:10.1038/nrneph.2016.51 |
Lu X K, Chen X G, Mu M, Wang J J, Wang X G, Wang D L, Yin Z J, Fan W L, Wang S, Guo L X, Ye W W. 2016. Genome-wide analysis of long noncoding RNAs and their responses to drought stress in cotton (Gossypium hirsutum L.). PLoS One, 11(6): e0156723.
DOI:10.1371/journal.pone.0156723 |
Ma Y L, Yang Y Z, Wang F, Moyer M P, Wei Q, Zhang P, Yang Z, Liu W J, Zhang H Z, Chen N W, Wang H, Wang H M, Qin H L. 2016. Long non-coding RNA CCAL regulates colorectal cancer progression by activating Wnt/β-catenin signalling pathway via suppression of activator protein 2α. Gut, 65(9): 1 494-1 504.
DOI:10.1136/gutjnl-2014-308392 |
Philippen L E, Dirkx E, da Costa-Martins P A, De Windt L J. 2015. Non-coding RNA in control of gene regulatory programs in cardiac development and disease. Journal of Molecular and Cellular Cardiology, 89: 51-58.
DOI:10.1016/j.yjmcc.2015.03.014 |
Roberts T C, Morris K V, Wood M J A. 2014. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philosophical Transactions of the Royal Society B:Biological Science, 369(1652): pii:20130507.
DOI:10.1098/rstb.2013.0507 |
Santosh B, Varshney A, Yadava P K. 2015. Non-coding RNAs:biological functions and applications. Cell Biochemistry& Function, 33(1): 14-22.
|
Sauvageau M, Goff L A, Lodato S, Bonev B, Groff A F, Gerhardinger C, Sanchez-Gomez D B, Hacisuleyman E, Li E, Spence M, Liapis S C, Mallard W, Morse M, Swerdel M R, D'Ecclessis M F, Moore J C, Lai V, Gong G C, Yancopoulos G D, Frendewey D, Kellis M, Hart R P, Valenzuela D M, Arlotta P, Rinn J L. 2013. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife, 2: e01749.
DOI:10.7554/eLife.01749 |
Shao C W, Li Q Y, Chen S L, Zhang P, Lian J M, Hu Q M, Sun B, Jin L J, Liu S S, Wang Z J, Zhao H M, Jin Z H, Liang Z, Li Y Z, Zheng Q M, Zhang Y, Wang J, Zhang G J. 2014. Epigenetic modification and inheritance in sexual reversal of fish. Genome Research, 24(4): 604-615.
DOI:10.1101/gr.162172.113 |
Sun L, Luo H T, Bu D C, Zhao G G, Yu K T, Zhang C H, Liu Y N, Chen R S, Zhao Y. 2013. Utilizing sequence intrinsic composition to classify protein-coding and long noncoding transcripts. Nucleic Acids Research, 41(17): e166.
DOI:10.1093/nar/gkt646 |
Sun M, Kraus W L. 2015. From discovery to function:the expanding roles of long noncoding RNAs in physiology and disease. Endocrine Review, 36(1): 25-64.
DOI:10.1210/er.2014-1034 |
Sun Y Y, Yu H Y, Zhang Q Q, Qi J, Zhong Q W, Chen Y J, Li C M. 2010. Molecular characterization and expression pattern of two zona pellucida genes in half-smooth tongue sole (Cynoglossus semilaevis). Comparative Biochemistry and Physiology Part B:Biochemistry and Molecular Biology, 155(3): 316-321.
DOI:10.1016/j.cbpb.2009.11.016 |
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley D R, Pimentel H, Salzberg S L, Rinn J L, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3): 562-578.
DOI:10.1038/nprot.2012.016 |
Viereck J, Kumarswamy R, Thum T. 2015. Long noncoding RNAs as inducers and terminators of vascular development. Circulation, 131(14): 1 236-1 238.
DOI:10.1161/CIRCULATIONAHA.115.015775 |
Wang L G, Park H J, Dasari S, Wang S Q, Kocher J P, Li W. 2013. CPAT:coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Research, 41(6): e74.
DOI:10.1093/nar/gkt006 |
Xu W T, Li H L, Dong Z D, Cui Z K, Zhang N, Meng L, Zhu Y, Liu Y, Li Y Z, Guo H, Ma J L, Wei Z F, Zhang N W, Yang Y M, Chen S L. 2016. Ubiquitin ligase gene neurl3 plays a role in spermatogenesis of half-smooth tongue sole (Cynoglossus semilaevis) by regulating testis protein ubiquitination. Gene, 592(1): 215-220.
DOI:10.1016/j.gene.2016.07.062 |
Yan B, Yao J, Liu J Y, Li X M, Wang X Q, Li Y J, Tao Z F, Song Y C, Chen Q, Jiang Q. 2015. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circulation Research, 116(7): 1 143-1 156.
DOI:10.1161/CIRCRESAHA.116.305510 |
Yan H, Chen Y D, Zhou S, Li C, Gong G Y, Chen X J, Wang T Z, Chen S L, Sha Z X. 2016. Expression Profile Analysis of miR-221 and miR-222 in Different Tissues and Head Kidney Cells of Cynoglossus semilaevis, Following Pathogen Infection. Marine Biotechnology, 18(1): 37-48.
|
Yue F, Cheng Y, Breschi A, Vierstra J, Wu W S, Ryba T, Sandstrom R, Ma Z H, Davis C, Pope B D, Shen Y, Pervouchine D D, Djebali S, Thurman R E, Kaul R, Rynes E, Kirilusha A, Marinov G K, Williams B A, Trout D, Amrhein H, Fisher-Aylor K, Antoshechkin I, DeSalvo G, See L H, Fastuca M, Drenkow J, Zaleski C, Dobin A, Prieto P, Lagarde J, Bussotti G, Tanzer A, Denas O, Li K W, Bender M A, Zhang M H, Byron R, Groudine M T, McCleary D, Pham L, Ye Z, Kuan S, Edsall L, Wu Y C, Rasmussen M D, Bansal M S, Kellis M, Keller C A, Morrissey C S, Mishra T, Jain D, Dogan N, Harris R S, Cayting P, Kawli T, Boyle A P, Euskirchen G, Kundaje A, Lin S, Lin Y, Jansen C, Malladi V S, Cline M S, Erickson D T, Kirkup V M, Learned K, Sloan C A, Rosenbloom K R, De Sousa B L, Beal K, Pignatelli M, Flicek P, Lian J, Kahveci T, Lee D, Kent W J, Santos M R, Herrero J, Notredame C, Johnson A, Vong S, Lee K, Bates D, Neri F, Diegel M, Canfield T, Sabo P J, Wilken M S, Reh T A, Giste E, Shafer A, Kutyavin T, Haugen E, Dunn D, Reynolds A P, Neph S, Humbert R, Hansen R S, De Bruijn M, Selleri L, Rudensky A, Josefowicz S, Samstein R, Eichler E E, Orkin S H, Levasseur D, Papayannopoulou T, Chang K H, Skoultchi A, Gosh S, Disteche C, Treuting P, Wang Y L, Weiss M J, Blobel G A, Cao X Y, Zhong S, Wang T, Good P J, Lowdon R F, Adams L B, Zhou X Q, Pazin M J, Feingold E A, Wold B, Taylor J, Mortazavi A, Weissman S M, Stamatoyannopoulos J A, Snyder M P, Guigo R, Gingeras T R, Gilbert D M, Hardison R C, Beer M A, Ren B, The Mouse ENCODE Consortium. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature, 515(7527): 355-364.
DOI:10.1038/nature13992 |