 2020, Vol. 38
2020, Vol. 38Institute of Oceanology, Chinese Academy of Sciences
Article Information
- ZHANG Bo, WU Yingying, WANG Xin, JIANG Wei, YIN Jianping, LIN Qiang
- Comparative analysis of mitochondrial genome of a deepsea crab Chaceon granulates reveals positive selection and novel genetic features
- Journal of Oceanology and Limnology, 38(2): 427-437
- http://dx.doi.org/10.1007/s00343-019-8364-x
Article History
- Received Dec. 18, 2018
- accepted in principle Apr. 4, 2019
- accepted for publication Jun. 12, 2019




2 Institution of South China Sea Ecology and Environmental Engineering, Chinese Academy of Sciences, Guangzhou 510275, China;
3 University of Chinese Academy of Sciences, Beijing 100049, China;
4 Beijing Advanced Sciences and Innovation Center of Chinese Academy of Sciences, Beijing 100049, China;
5 Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China
The deep-sea is the most extensive ecosystem on earth (Rex, 1981). Unlike terrestrial and shallow organisms, deep-sea organisms survive in an extremely harsh environment, which including hundreds of bars of pressure, low oxygen, scarce food, constant darkness, and low temperatures (Sanders and Hessler, 1969). Because of the sparse animal life and technical difficulties in sampling the deep-sea benthos, most knowledge regarding deepsea organisms is restricted to marine microbes and morphological distinctions of a few animal species. There is little information regarding the adaptive genetic mechanisms in the large deep-sea organisms (Etter et al., 1999; Sogin et al., 2006; Jebbar et al., 2015; Zhang et al., 2015; Coscia et al., 2018).
Mitochondria is one of the most essential organelle in eukaryotic cells (Bernt et al., 2013), and plays an important role in energy metabolism and various biosynthetic pathways (Green and Reed, 1998; Newmeyer and Ferguson-Miller, 2003). Mitochondrial DNA is more strongly influenced by evolutionary processes than nuclear DNA largely because it has a smaller effective population size and does not undergo recombination. The mitochondrial genome (mitogenome) has been widely used for study in genetic diversity, phylogeography, phylogenetic relationships, and adaptive mechanisms (Da et al., 2008; Hassanin et al., 2009; Yu et al., 2011; Jin et al., 2015; Liao et al., 2016). Recently, several mitochondrial genes have shown significantly adaptive evolution, including the cytochrome b gene of alpacas (Da et al., 2008), the cytochrome c oxidase gene of plateau pikas (Luo et al., 2008), the NADH dehydrogenase 6 gene of domestic horses (Ning et al., 2010), and the ATP synthase genes of Caprinae (Hassanin et al., 2009).
Crabs are typical benthic organisms and distribute all over the world. To adapt to different environments, they have evolved a broad range of phenotypes. Thus, it can be used as an index for the study of adaptive mechanisms to some local environment. The mitogenomes of crabs are typically closed circular molecules, with 14 kb to 18 kb in length. They encode the following 37 genes: 13 protein-coding genes (PCGs), 22 transfer RNA (tNRA) genes, 2 ribosomal RNA (rRNA) genes, and a putative control region (CR) (Liu and Cui, 2010; Ma et al., 2013; Tang et al., 2017). However, the basic genetic information of crabs under different environments is far from enough, especially for extremely environment. To date, Chaceon granulates (NC_AB769383) was sequenced, but the genomic characteristics have not been illuminated in detail. In this study, the complete mitogenome of another deep-sea C. granulates was sequenced. Furthermore, in comparison with those of shallow water crabs, some unique genomic features were characterized. Interestingly, some novel genetic features that could be involved in the adaptation of deep-sea environment were identified. This work made up the data of crab genome, and should be useful for studies on crab evolution and adaptive mechanisms.
2 METERIAL AND METHOD 2.1 Specimens and DNA extractionThe C. granulates specimen was collected by deep-water research vessel on December 15, 2014, on the Yap seamount (137.46°E; 8.52°N) in the Pacific Ocean at 477 m depth. The specimen was not a member of an endangered or protected species, and no specific permits were required. The specimen was stored in 99% ethanol and kept at 4℃. DNA was extracted using a Genomic DNA Kit (Tiangen Co., Beijing, China) according to the manufacturer's instructions.
2.2 PCR and sequencingThe complete mitogenome was amplified by using overlapping long-PCR. Five pairs of primers were designed at conserved regions of crab mitogenome (Supplementary Table S1). All PCR reactions were performed in 50 μL volume, with 1 μL of template DNA (approximately 100 ng), 0.3 μmol/L of each primer, 5 μL of 10 × LA Taq buffer (Mg2+ plus), 5 μL of dNTP Mix (2.5 mmol/L), and 1 U of LA Taq (TaKaRa, Japan). The PCR amplifications were performed according to the following procedure: one cycle of denaturation for 5 min at 94℃, 30 cycles of 40 s at 94℃, 40 s at the primer-specific annealing temperature, 5 min at 72℃, and finally a 10-min extension at 72℃. After purification (TIANgel Midi Purification Kit DP209, Beijing), the PCR products were directly sequenced in both directions by using the PCR primers. Sequencing was performed by Thermo Fisher Scientific (Guangzhou, China).
2.3 Sequence assembly and annotationThe sequence alignments were conducted using
Clustal X. The PCGs and rRNA genes were identified
with BLAST and the NCBI database, and were
compared with the mitogenome sequences of several
other crab species (Supplementary Table S2). The
tRNA genes and their secondary structures were
predicted by using the web-based tRNAscan-SE 1.21
(Lowe and Eddy, 1997). The tRNASer, which was not
found by using the software tools, was identified on
the basis of sequence similarity to the published crab
mitochondrial tRNASer (Ma et al., 2015; Miller et al., 2005). The skew in the nucleotide composition was
calculated with AT-skew and GC-skew and measured
according to the following formulae: AT-skew=(A–T)/
(A+T) and GC-skew=(G–C)/(G+C) (
The selective pressure on the crab mitogenome was evaluated using CODEML in the PAML package. Two different tree-building methods were used because the CODEML likelihood analysis is sensitive to the tree-topology. The two-ratio and free-ratio model (M1 model) was used for the mitogenome analysis. The branch-site model was used to determine whether these genes have undergone positive selection on foreground branches. Bayes Empirical Bayes (BEB) analysis was used to calculate the Bayesian posterior probability of the positively selected sites.
2.5 Phylogenetic analysisTo illustrate the phylogenetic relationships among crabs, the complete mitogenomes of 23 Decapoda species were downloaded from the GenBank database (Supplementary Table S2). Harpiosquilla harpax (Stomatopoda) was selected as an outgroup. The concatenated nucleotide sequences of 13 energy pathway PCGs were aligned using Clustal X with the default settings. The maximum likelihood (ML) method was used to analyze the phylogenetic trees. The GTR+I+G model was selected as the best nucleotide substitution model by using ModelTest 3.7 (Posada and Crandall, 1998). The ML analysis was performed with MEGA 5.1 with 1 000 bootstrap replicates.
3 RESULT AND DISCUSSION 3.1 Genome organizationThe mitochondrial DNA of the crab is a circular molecule, with 16 126 bp in length. The sequence alignment showed the most identical (98%) with another C. granulates (NC_AB769383) uploaded before. Like most other metazoan mitogenomes (Green and Reed, 1998; Boore, 1999; Bernt et al., 2013), the present crab mitogenome contains 13 PCGs, 22 tRNA genes, 2 rRNA genes, and a control region (CR) (Table 1, Fig. 1). Of the 37 genes, 23 genes were encoded by the heavy strand (H-strand). The gene order and orientation were identical to other decapods (Yamauchi et al., 2003; Miller et al., 2005; Liu and Cui, 2010; Ma et al., 2013). Eight gene overlaps and eleven intergenic spacers were observed. Except for a 527-bp untranslated region between tRNAGlu and tRNAHis, the gene overlaps and intergenic spacers were similar to those in other sequenced Decapod crabs. The complete mitochondrial DNA sequence was deposited in the GenBank database under the accession number KU507298.

|
| Fig.1 Graphical map of the complete mitogenome of C. granulates Different genes were represented by differently colored boxes. tRNAs were displayed according in one-letter code. Genes encoded by the H-strand were showed outside the circle, and those encoded by the L-strand were showed inside the circle. The direction of the arrows showed the direction of transcription. The inner ring showed the GC content in the mitogenome. |
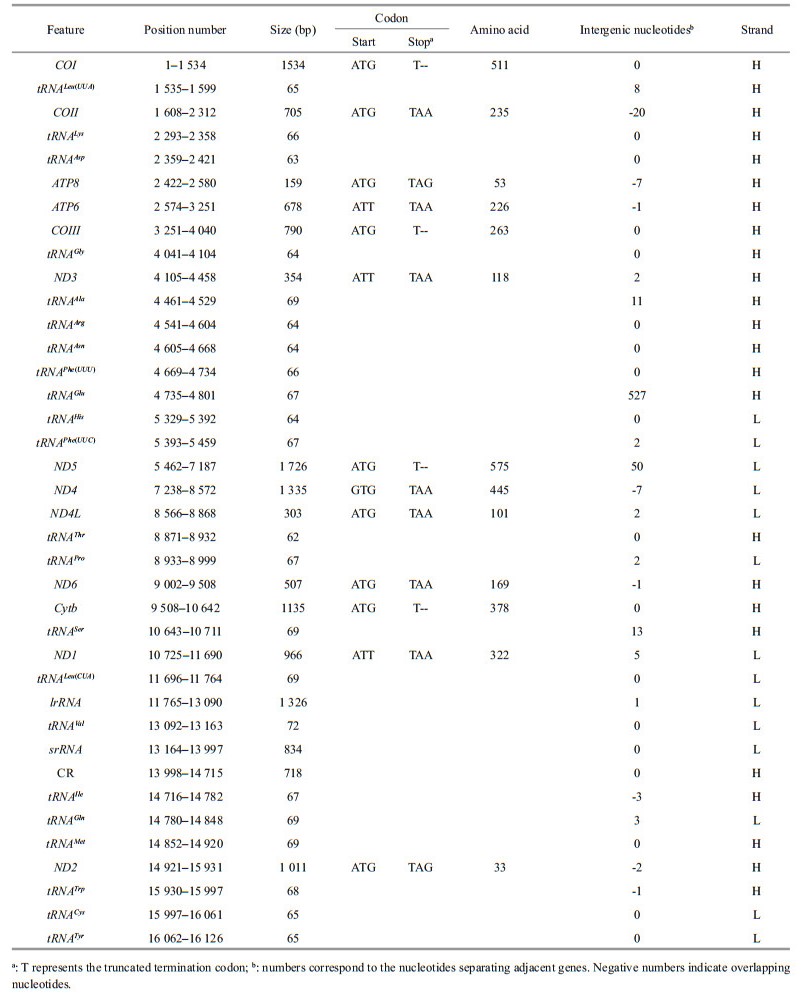
The metazoan mitogenome usually has a strand specific bias in nucleotide composition (Hassanin et al., 2005). In the present study, the nucleotide composition had a bias toward A and T. The overall A+T content in the H-strand was 69.19% (Supplementary Table S3), which is within the range of the rates observed in other Decapods (Shen et al., 200). In C. granulates mitogenome, similarly to other crabs (Liu and Cui, 2010; Ma et al., 2013), the highest A+T content was detected in the CR (77.58%), and the lowest A+T content was found in the 13 PCGs (67.03%). The AT-skew and GC-skew showed a similar tendency in decapods (Liu and Cui, 2010; Ma et al., 2013). The AT-skew (-0.039) and the GC-skew (-0.205) in the H-strand were negative, thus indicating a preference for A and C in C. granulates mitogenome.
3.2 Protein-coding genesIn total, 13 PCGs were identified in the C. granulates mitogenome (Table 1). These genes spanned 11 204 bp and encoded 3 724 amino acids. The arrangements of PCGs in several Brachyura species were consistent (Fig. 2). In C. granulates mitogenome 9 PCGs were encoded by the H-strand (Fig. 1). Except for ND4, which was initiated by a rare initiation codon (GTG), the remaining genes started with the typical ATN codons (ATG or ATT). ATG was the most common initiation codon, which initiated 9 of the 13 PCGs (Table 1). Three kinds of termination codons (TAA, TAG, and an incomplete stop codon T) were observed. The most common termination codon was TAA. In the metazoan mitogenome, incomplete termination codons produce functional termination codons through polycistronic transcriptional cleavage (Ojala et al., 1981; Boore, 2001). In the present study, 4 PCGs were terminated by the truncated termination codon T, in which the missing nucleotides may be produced through post-transcriptional polyadenylation (Ojala et al., 1981).

|
| Fig.2 Linearized schemes of the mitochondrial gene arrangements in Brachyura C. destructor (Astacidea as an outgroup) Gene lengths correspond to the relative lengths of the genomes. tRNAs were displayed according to the one-letter code. Species names and NCBI accession numbers were provided under each linearized scheme. |
The codon usage in the 13 PCGs revealed that UUA (Leu), AUU (Ile), and UUU (Phe) were the three most frequently utilized codons in C. granulates mitogenome (Supplementary Table S4, Fig. 3a). The codon distribution patterns in 11 Brachyura and 1 Anomura species were highly conserved (Fig. 3b).

|
| Fig.3 Codon usage in crabs a. codon distribution in C. granulates mitogenome; b. comparison of mitogenomic codon usage among 12 crabs. |
The selective pressures imposed on the crab mitogenomes were evaluated using CodeML in the PAML package (Table 2). The result showed that the mitochondrial sequences in the genus Chaceon experienced different selective pressures compared with shallow species, and significant evidence of positive selection was detected in two sites (33S in ND3 and 502I in ND5) of C. granulates (BEB value > 0.95).
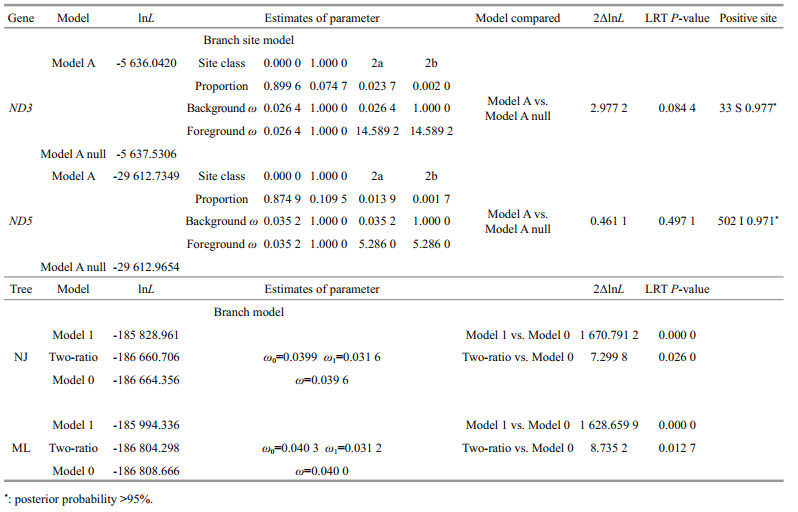
During the evolution, protein positive selection may act in very short episodes, and the effect may be only on a few sites along lineages in the phylogeny (Sun et al., 2018). The severe environmental conditions can affect the metabolism and direct selection of mitochondrial DNA (Ning et al., 2010). As the key components of energy metabolism and various biosynthetic pathways (Green and Reed, 1998; Newmeyer and Ferguson-Miller, 2003), the mutations in mitochondrial protein-coding genes may influence the electron transport chain and further influence the energy metabolism and other biosynthetic pathways (Sun et al., 2018). Many studies have recently shown evidence for positive selection acting on the mitochondrial genome, emphasizing its potential role in adaptive divergence and speciation (Jacobsen et al., 2016). Organisms living under extreme environmental conditions, there must be certain modifications in energy metabolism to adapt to environment (Zhang et al., 2017a, b; Sun et al., 2018). As a proton pump, the NADH dehydrogenase complex is the first and the largest enzyme complex in the respiratory chain. Thus, it plays an important role on the adaptive evolution in many species. In Alvinocarididae lineages, the residues with the highest number of positively selected sites are within nad1–5 (Sun et al., 2018), and is considered associated with deep sea hydrothermal vents adaptation. ND2 and ND6 were found to be under positive selection in mitogenome analysis of Chinese snub-nosed monkeys, it was known to be related to adaptive changes high altitude and cold weather stress (Yu et al., 2011). In Tibetan horses, ND6 was found to be under positive selection, which was associated with high altitude living adaptation (Ning et al., 2010). In whitefish (Coregonus spp.), ND2 showed a highly elevated dN/dS ratio, which was considered to drive the adaptive evolution (Jacobsen et al., 2016). The mitochondrial whole-genome comparison study in 40 Tibetan and 50 Han Chinese people provide clues for the existence of adaptive selection for the ND2 in Tibetans, which likely contributed to adaptation to their specific geographic environment, such as high altitude (Gu et al., 2012).
In this study, two sites of NADH dehydrogenase complex presented significant evidence of positive selection. The result supports the hypothesis of adaptive evolution in the mitogenome of deep-sea environment. The mutations in these subunits may influence the efficiency of the proton-pumping process. They may potentially have functional implications to the energy metabolism. Considering the living environment, the mutations in NADH dehydrogenase complex may contribute to the adaptation to deep-sea environment.
3.4 tRNA genes and rRNA genesIn total, 22 tRNAs were identified in C. granulates (Table 1), with sizes ranging from 62 to 72 bp. Of the 22 tRNA genes, 14 tRNA genes were located on the H-strand. Except for tRNASer(AGA), all 21 tRNAs folded into a cloverleaf secondary structure (Supplementary Fig.S1). The tRNASer(AGA) presented an unusual secondary structure lacking the stem-loop structure in the DHU arm, which is observed in other crabs (Liu and Cui, 2010). In addition, 4 unmatched base pairs were observed in the C. granulates mitogenome. The tRNACys contained an A-A mismatch on the DHU arm, but the remaining 3 mismatched tRNAs occurred in the amino acid acceptor arm. Such stem mismatches appear to be a common phenomenon in mitochondrial tRNAs in many species (Miller et al., 2005; Liao et al., 2010; Jiang et al., 2013; Wang et al., 2016) and are probably corrected through a post RNA-editing mechanism (Lavrov et al., 2000).
Like most metazoan mitogenomes, 2 rRNA genes (lrRNA and srRNA) were present in C. granulates (Table 1). In C. granulates mitogenome, the lrRNA and srRNA genes were located on the L-strand and contained 1 326 bp and 834 bp, respectively. The A+T contents were 74.53% and 73.50%. The lsRNA gene was located between the tRNALeu(CUA) and tRNAVal genes. The srRNA gene was located between the tRNAVal gene and the putative CR.
3.5 Non-coding regionsThirteen non-coding regions were identified in the mitogenome of C. granulates (Table 1). The longest intergenic region was the putative CR. It is considered essential for mitochondrial genes transcription and replication in vertebrates (Fernández-Silva et al., 2003). It is usually considered the most variable portion of the mitogenome (Marshall and Baker, 1997). The nucleotide composition of the CR (H-strand) was 314 A (43.73%), 243 T (33.84%), 103 C (14.35%), and 58 G (8.08%). It showed a similar tendency in Brachyuran crabs. However, the nucleotide composition and length variations were evident among crabs. The nucleotide alignment of the CR in Brachyuran crabs showed low homology, which was also confirmed in Charybdis japonica (Liu and Cui, 2010). The length of the CRs in crabs were range from 514 bp to 1 435 bp (Supplementary Table S3). The result is consistent with the studies in other crustaceans (Valverde et al., 1994; Umetsu et al., 2002).
In addition to the largest CR (718 bp) in the mitogenome of C. granulates, another non-coding sequence (527 bp) was detected, which is a unique insertion that just detected in Chaceon crabs. Moreover, the sequence analysis of the 527 bp noncoding sequence exhibited several typical "CR-like" characteristics. Firstly, similar to the defined CR, the non-coding sequence was much larger than the remaining 11 non-coding regions, which ranged from 1 bp to 50 bp. The result is identical to the CR observed in Pocillopora (Flot and Tillier, 2007). Secondly, both of the 718-bp CR (H-strand) and the 527-bp genetic fragment (L-strand) showed similar nucleotide composition. The nucleotide composition of the 527-bp non-coding region was 229 A (43.45%), 172 T (32.64%), 45 C (8.54%), and 81 G (15.37%). Both parts showed almost the same A+T content rate, which is higher than that in the other regions in the mitochondrial DNA. High rate of A+T is a common feature in all organisms except primates (Sbisa et al., 1997). Thirdly, although the CR in Brachyuran crabs showed low homology; several conserved motifs were identified in both parts and other several CRs of Brachyuran crab (Supplementary Fig.S2c). The "TACAT" motif, which is observed in some fish (Guo et al., 2003), was found in C. granulates (Supplementary Fig.S2a & b). The "G(A)nT" motif, which was present in the 3′ flanking sequences of mammalian, amphibian, and fish mitochondrial L-strand replication origins, and showed an universal conservation and functional importance related to replication origins (Zhang et al., 1995), was also detected in the 527-bp non-coding region (Supplementary Fig.S2a & b). Thus, we presumed that the 527-bp non-coding region could be a regulatory element in the present study.
Further structural analyses showed that the secondary structure of the 527-bp non-coding region presented several stem-loop structures (Supplementary Fig.S3), which is a characteristic feature of the origin of light-strand replication (OL) in vertebrates (Clayton, 1991). Moreover, the locus of the 527-bp non-coding region in mitogenome was identical to the OL in most vertebrates. In most vertebrates, the OL was located in a cluster of tRNA genes (between tRNAAsn and tRNACys), which known as the WANCY region (Kawaguchi et al., 2001; Jin et al., 2015). Therefore, we concluded the 527-bp non-coding region could be the OL of C. granulates. At present, among Brachyuran crabs, such non-coding region (OL) was detected in mitogenomes of deep-sea Chaceon crabs only (Fig. 2).
In the study of deep-sea hydrothermal vents and cold seeps Alvinocaris longirostris, seven putatively duplicated gene clusters of cytochrome P450s were expressed differentially, which is considered to contribute to the adaptation to harsh conditions (Hui et al., 2018). Considering the OL can influence the replication and transcription of mitochondrial genes, we speculated the OL could participate in the regulation of mitochondrial genes expression. Thus, it could be indirectly involved in adjusting mitochondrial energy metabolism to adapt to the deep-sea environment.
3.6 Phylogenetic analysesWe performed a phylogenetic analysis of the crabs based on the nucleotide datasets of 13 mitochondrial energy pathway PCGs (Fig. 4). The complete mitochondrial genome of C. granulates provides well resolved molecular phylogeny of the Decapoda. The phylogenetic tree supported the hypothesis that Decapoda was reorganized into the Pleocyemata and Dendrobranchiata suborders (Burkenroad, 1981), a result consistent with the decapod phylogeny based on other molecular data (Tsang et al., 2008). Together with 11 other species, C. granulates first formed a monophyletic group with a 100% bootstrapping value, which clustered in the Brachyura clade. Anomura, Achelata, Astacidea and Caridea composed the other clades in the Pleocyemata group. The Brachyura clade showed a close relationship with the Anomura clade, as has been shown in another study (Tsang et al., 2008; Liu and Cui, 2010; Ma et al., 2015).

|
| Fig.4 Phylogenetic analysis Phylogenetic tree of species in Decapoda. Harpiosquilla harpax was selected as outgroup. Bootstrap support values were showed on the nodes. |
The complete mitogenome of a deep-sea crab, C. granulates, was characterized and compared with other shallow decapods. The complete mitogenome of C. granulates is a typical circular molecule, with 16 126 bp in length. The genetic composition, order, and orientation are similar to those of closely related crabs, belonging to the Brachyura branch clade of Decapoda. Several genes showed strong evidence of positive selection, in genus Chaceon of the mitogenomic analysis, and ND3 and ND5 were determined to be under positive selection with a BEB value >95%, by using the branch-site model in CODEML. C. granulates mitogenome possesses a unique "OL" that is not present in shallow crabs. Therefore, given that the OL in mitogenomes is essential for mitochondrial genes expressional regulation, as well as positive selection genes, we speculated that two special gene features might play important roles in regulating the mitochondrial energy metabolism that could involve in the adaptation to the deep-sea conditions. The data presented in this study may shed a light on the knowledge of mitogenomic adaptation in the deep-sea environment.
5 DATA AVAILABILITY STATEMENTThe authors declare that all data supporting the findings of this study are available within the article and its supplementary files.
Bernt M, Braband A, Schierwater B, Stadler P F. 2013. Genetic aspects of mitochondrial genome evolution. Molecular Phylogenetics and Evolution, 69(2): 328-338.
DOI:10.1016/j.ympev.2012.10.020 |
Boore J L. 1999. Animal mitochondrial genomes. Nucleic Acids Research, 27(8): 1 767-1 780.
DOI:10.1093/nar/27.8.1767 |
Boore J L. 2001. Complete mitochondrial genome sequence of the polychaete annelid Platynereis dumerilii. Molecular Biology and Evolution, 18(7): 1 413-1 416.
DOI:10.1093/oxfordjournals.molbev.a003925 |
Burkenroad M D. 1981. The higher taxonomy and evolution of Decapoda (Crustacea). Transactions of the San Diego Society of Natural History, 19(17): 251-268.
|
Clayton D A. 1991. Replication and transcription of vertebrate mitochondrial-DNA. Annual Review of Cell Biology, 7(1): 453-478.
DOI:10.1146/annurev.cb.07.110191.002321 |
Coscia I, Castilho R, Massa-Gallucci A, Sacchi C, Cunha R L, Stefanni S, Helyar S J, Knutsen H, Mariani S. 2018. Genetic homogeneity in the deep-sea grenadier Macrourus berglax across the North Atlantic Ocean. Deep Sea Research Part I:Oceanographic Research Papers, 132: 60-67.
DOI:10.1016/j.dsr.2017.12.001 |
Da Fonseca R R, Johnson W E, O'Brien S J, Ramos M J, Antunes A. 2008. The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics, 9: 119.
DOI:10.1186/1471-2164-9-119 |
Etter R J, Rex M A, Chase M C, Quattro J M. 1999. A genetic dimension to deep-sea biodiversity. Deep Sea Research Part I:Oceanographic Research Papers, 46(6): 1095-1 099.
DOI:10.1016/S0967-0637(98)00100-9 |
Fernández-Silva P, Enriquez J A, Montoya J. 2003. Replication and transcription of mammalian mitochondrial DNA. Experimental Physiology, 88(1): 41-56.
|
Flot J F, Tillier S. 2007. The mitochondrial genome of Pocillopora (Cnidaria:Scleractinia) contains two variable regions:The putative D-loop and a novel ORF of unknown function. Gene, 401(1-2): 80-87.
DOI:10.1016/j.gene.2007.07.006 |
Green D R, Reed J C. 1998. Mitochondria and apoptosis. Science, 281(5381): 1309-1 312.
DOI:10.1126/science.281.5381.1309 |
Gu M L, Dong X Q, Shi L, Shi L, Lin K Q, Huang X Q, Chu J Y. 2012. Differences in mtDNA whole sequence between Tibetan and Han populations suggesting adaptive selection to high altitude. Gene, 496(1): 37-44.
DOI:10.1016/j.gene.2011.12.016 |
Guo X H, Liu S J, Liu Y. 2003. Comparative analysis of the mitochondrial DNA control region in cyprinids with different ploidy level. Aquaculture, 224(1-4): 25-38.
DOI:10.1016/S0044-8486(03)00168-6 |
Hassanin A, Léger N, Deutsch J. 2005. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Systematic Biology, 54(2): 277-298.
|
Hassanin A, Ropiquet A, Couloux A, Cruaud C. 2009. Evolution of the mitochondrial genome in mammals living at high altitude:new insights from a study of the Tribe Caprini (Bovidae, Antilopinae). Journal of Molecular Evolution, 68(4): 293-310.
DOI:10.1007/s00239-009-9208-7 |
Hui M, Cheng J, Sha Z L. 2018. Adaptation to the deep-sea hydrothermal vents and cold seeps:Insights from the transcriptomes of Alvinocaris longirostris in both environments. Deep Sea Research Part I:Oceanographic Research Papers, 135: 23-33.
DOI:10.1016/j.dsr.2018.03.014 |
Jacobsen M W, Da Fonseca R R, Bernatchez L, Hansen M M. 2016. Comparative analysis of complete mitochondrial genomes suggests that relaxed purifying selection is driving high nonsynonymous evolutionary rate of the NADH2 gene in whitefish (Coregonus ssp).. Molecular Phylogenetics and Evolution, 95: 161-170.
DOI:10.1016/j.ympev.2015.11.008 |
Jebbar M, Franzetti B, Girard E, Oger P. 2015. Microbial diversity and adaptation to high hydrostatic pressure in deep-sea hydrothermal vents prokaryotes. Extremophiles, 19(4): 721-740.
DOI:10.1007/s00792-015-0760-3 |
Jiang L C, Wang G C, Tan S, Gong S A, Yang M, Peng Q K, Peng R, Zou F D. 2013. The complete mitochondrial genome sequence analysis of Tibetan argali (Ovis ammon hodgsoni):implications of Tibetan argali and Gansu argali as the same subspecies. Gene, 521(1): 24-31.
DOI:10.1016/j.gene.2013.03.049 |
Jin X X, Wang R X, Wei T, Tang D, Xu T J. 2015. Complete mitochondrial genome sequence of Tridentiger bifasciatus and Tridentiger barbatus (Perciformes, Gobiidae):a mitogenomic perspective on the phylogenetic relationships of Gobiidae. Molecular Biology Reports, 42(1): 253-265.
DOI:10.1007/s11033-014-3768-3 |
Kawaguchi A, Miya M, Nishida M. 2001. Complete mitochondrial DNA sequence of Aulopus japonicus(Teleostei:Aulopiformes), a basal Eurypterygii:longer DNA sequences and higher-level relationships. Ichthyological Research, 48(3): 213-223.
DOI:10.1007/s10228-001-8139-0 |
Lavrov D V, Brown W M, Boore J L. 2000. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proceedings of the National Academy of Sciences of the United States of America, 97(25): 13738-13 742.
DOI:10.1073/pnas.250402997 |
Liao F, Wang L, Wu S, Li Y P, Zhao L, Huang G M, Niu C J, Liu Y Q, Li M G. 2010. The complete mitochondrial genome of the fall webworm, Hyphantria cunea(Lepidoptera:Arctiidae). International Journal of Biological Sciences, 6(2): 172-186.
|
Liao Y Y, Mo G D, Sun J L, Wei F Y, Liao D J. 2016. Genetic diversity of Guangxi chicken breeds assessed with microsatellites and the mitochondrial DNA D-loop region. Molecular Biology Reports, 43(5): 415-425.
DOI:10.1007/s11033-016-3976-0 |
Liu Y, Cui Z X. 2010. Complete mitochondrial genome of the Asian paddle crab Charybdis japonica (Crustacea:Decapoda:Portunidae):gene rearrangement of the marine brachyurans and phylogenetic considerations of the decapods. Molecular Biology Reports, 37(5): 2 559-2 569.
DOI:10.1007/s11033-009-9773-2 |
Lowe T M, Eddy S R. 1997. tRNAscan-SE:A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research, 25(5): 955-964.
DOI:10.1093/nar/25.5.955 |
Luo Y J, Gao W X, Gao Y Q, Tang S, Huang Q Y, Tan X L, Chen J, Huang T S. 2008. Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion, 8(5-6): 352-357.
DOI:10.1016/j.mito.2008.07.005 |
Ma H Y, Ma C Y, Li C H, Lu J X, Zou X, Gong Y Y, Wang W, Chen W, Ma L B, Xia L J. 2015. First mitochondrial genome for the red crab (Charybdis feriata) with implication of phylogenomics and population genetics. Scientific Reports, 5: 11 524.
|
Ma H Y, Ma C Y, Li X C, Xu Z, Feng N N, Ma L B. 2013. The complete mitochondrial genome sequence and gene organization of the mud crab (Scylla paramamosain) with phylogenetic consideration. Gene, 519(1): 120-127.
DOI:10.1016/j.gene.2013.01.028 |
Marshall H D, Baker A J. 1997. Structural conservation and variation in the mitochondrial control region of fringilline finches (Fringilla spp) and the greenfinch (Carduelis chloris). Molecular Biology and Evolution, 14(2): 173-184.
DOI:10.1093/oxfordjournals.molbev.a025750 |
Miller A D, Murphy N P, Burridge C P, Austin C M. 2005. Complete mitochondrial DNA sequences of the decapod crustaceans Pseudocarcinus gigas (Menippidae) and Macrobrachium rosenbergii (Palaemonidae). Marine Biotechnology, 7(4): 339-349.
DOI:10.1007/s10126-004-4077-8 |
Newmeyer D D, Ferguson-Miller S. 2003. Mitochondria:releasing power for life and unleashing the machineries of death. Cell, 112(4): 481-490.
DOI:10.1016/S0092-8674(03)00116-8 |
Ning T, Xiao H, Li J, Hua S, Zhang Y P. 2010. Adaptive evolution of the mitochondrial ND6 gene in the domestic horse. Genetics and Molecular Research, 9(1): 144-150.
DOI:10.4238/vol9-1gmr705 |
Ojala D, Montoya J, Attardi G. 1981. tRNA punctuation model of RNA processing in human mitochondria. Nature, 290(5806): 470-474.
DOI:10.1038/290470a0 |
Perna N T, Kocher T D. 1995. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. Journal of Molecular Evolution, 41(3): 353-358.
DOI:10.1007/BF01215182 |
Posada D, Crandall K A. 1998. MODELTEST:testing the model of DNA substitution. Bioinformatics, 14(9): 817-818.
DOI:10.1093/bioinformatics/14.9.817 |
Rex M A. 1981. Community structure in the deep-sea Benthos. Annual Review of Ecology and Systematics, 12(1): 331-353.
DOI:10.1146/annurev.es.12.110181.001555 |
Sanders H L, Hessler R R. 1969. Ecology of the deep-sea benthos. Science, 163(3874): 1 419-1 424.
DOI:10.1126/science.163.3874.1419 |
Sbisa E, Tanzariello F, Reyes A, Pesole G, Saccone C. 1997. Mammalian mitochondrial D-loop region structural analysis:identification of new conserved sequences and their functional and evolutionary implications. Gene, 205(1-2): 125-140.
DOI:10.1016/S0378-1119(97)00404-6 |
Shen X, Ren J F, Cui Z X, Sha Z L, Wang B, Xiang J H, Liu B. 2007. The complete mitochondrial genomes of two common shrimps (Litopenaeus vannamei and Fenneropenaeus chinensis) and their phylogenomic considerations. Gene, 403(1-2): 98-109.
DOI:10.1016/j.gene.2007.06.021 |
Sogin M L, Morrison H G, Huber J A, Welch D M, Huse S M, Neal P R, Arrieta J M, Herndl G J. 2006. Microbial diversity in the deep sea and the underexplored "rare biosphere". Proceedings of the National Academy of Sciences of the United States of America, 103(32): 12 115-12 120.
DOI:10.1073/pnas.0605127103 |
Sun S E, Hui M, Wang M X, Sha Z L. 2018. The complete mitochondrial genome of the alvinocaridid shrimp Shinkaicaris leurokolos (Decapoda, Caridea):insight into the mitochondrial genetic basis of deep-sea hydrothermal vent adaptation in the shrimp. Comparative Biochemistry and Physiology Part D:Genomics and Proteomics, 25: 42-52.
DOI:10.1016/j.cbd.2017.11.002 |
Tang B P, Liu Y, Xin Z Z, Zhang D Z, Wang Z F, Zhu X Y, Wang Y, Zhang H B, Zhou C L, Chai X Y, Liu Q N. 2017. Characterisation of the complete mitochondrial genome of Helice wuana (Grapsoidea:Varunidae) and comparison with other Brachyuran crabs. Genomics, 110(4): 221-230.
|
Tsang L M, Ma K Y, Ahyong S T, Chan T Y, Chu K H. 2008. Phylogeny of Decapoda using two nuclear protein-coding genes:origin and evolution of the Reptantia. Molecular Phylogenetics and Evolution, 48(1): 359-368.
DOI:10.1016/j.ympev.2008.04.009 |
Umetsu K, Iwabuchi N, Yuasa I, Saitou N, Clark P F, Boxshall G, Osawa M, Igarashi K. 2002. Complete mitochondrial DNA sequence of a tadpole shrimp (Triops cancriformis)and analysis of museum samples. Electrophoresis, 23(24): 4 080-4 084.
DOI:10.1002/elps.200290024 |
Valverde J R, Batuecas B, Moratilla C, Marco R, Garesse R. 1994. The complete mitochondrial DNA sequence of the crustacean Artemia franciscana. Journal of Molecular Evolution, 39(4): 400-408.
DOI:10.1007/BF00160272 |
Wang Z L, Li C, Fang W Y, Yu X P. 2016. The complete mitochondrial genome of two Tetragnatha spiders(Araneae:Tetragnathidae):severe truncation of tRNAs and novel gene rearrangements in Araneae. International Journal of Biological Sciences, 12(1): 109-119.
DOI:10.7150/ijbs.12358 |
Yamauchi M M, Miya M U, Nishida M. 2003. Complete mitochondrial DNA sequence of the swimming crab, Portunus trituberculatus (Crustacea:Decapoda:Brachyura). Gene, 311: 129-135.
DOI:10.1016/S0378-1119(03)00582-1 |
Yu L, Wang X P, Ting N, Zhang Y P. 2011. Mitogenomic analysis of Chinese snub-nosed monkeys:evidence of positive selection in NADH dehydrogenase genes in highaltitude adaptation. Mitochondrion, 11(3): 497-503.
DOI:10.1016/j.mito.2011.01.004 |
Zhang B, Zhang Y H, Wang X, Zhang H X, Lin Q. 2017a. The mitochondrial genome of a sea anemone Bolocera sp. exhibits novel genetic structures potentially involved in adaptation to the deep-sea environment. Ecology and Evolution, 7(13): 4 951-4 962.
|
Zhang D X, Szymura J M, Hewitt G M. 1995. Evolution and structural conservation of the control region of insect mitochondrial DNA. Journal of Molecular Evolution, 40(4): 382-391.
DOI:10.1007/BF00164024 |
Zhang Q L, Zhang L, Zhao T X, Wang J, Zhu Q H, Chen J Y, Yuan M L. 2017b. Gene sequence variations and expression patterns of mitochondrial genes are associated with the adaptive evolution of two Gynaephora species(Lepidoptera:Lymantriinae) living in different highelevation environments. Gene, 610: 148-155.
DOI:10.1016/j.gene.2017.02.014 |
Zhang Y, Li X G, Bartlett D H, Xiao X. 2015. Current developments in marine microbiology:high-pressure biotechnology and the genetic engineering of piezophiles. Current Opinion in Biotechnology, 33: 157-164.
DOI:10.1016/j.copbio.2015.02.013 |