 2020, Vol. 38
2020, Vol. 38Institute of Oceanology, Chinese Academy of Sciences
Article Information
- JIANG Fengjuan, YUE Xin, ZHANG Shujing, YU Jiajia, WANG Rui, LIU Baozhong, WANG Hongxia
- Heritability of resistance-related gene expression traits and their correlation with body size of clam Meretrix petechialis
- Journal of Oceanology and Limnology, 38(2): 571-578
- http://dx.doi.org/10.1007/s00343-019-8326-3
Article History
- Received Nov. 19, 2018
- accepted in principle Mar. 25, 2019
- accepted for publication Aug. 5, 2019




2 Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266000, China;
3 University of Chinese Academy of Sciences, Beijing 100049, China
Meretrix petechialis is one of the most important commercial clam species farmed in China (Liu et al., 2006). Despite continuous progress and improvement in artificial reproductive technologies and aquaculture techniques, clam diseases remain a major limiting factor in the clam culture industry. Outbreaks of vibriosis in clams often affect the profitability and sustainability of aquaculture operations. From a sustainability perspective, selective breeding for disease resistant clams is an attractive strategy for disease prevention (Liang et al., 2017). However, in contrast to many other economically important traits (e.g., growth rate, feed conversion efficiency), genetic improvement of disease resistance has been hindered by the difficulty of measuring appropriate phenotypes accurately (Bishop and Woolliams, 2014). Therefore, markers accurately reflecting disease resistance and having a significant genetic component are necessary in resistance selection.
To date, some association analyses between molecular markers and Vibrio resistance traits have been reported in M. petechialis (Nie et al., 2013, 2015; Zou and Liu, 2016). However, the effectiveness of these markers is minor, accounting for only a small fraction of the genetic variation (Yang et al., 2010). Thus, more attempts are needed to develop markers that are closely linked to Vibrio resistance. In previous studies, the use of gene expression profiles as indirect tests for improved disease resistance has been proposed for selective breeding in Atlantic salmon (Robinson and Hayes, 2008). Research on M. petechialis has demonstrated that hundreds of genes are involved in the physiological and immunological response to bacterial infection (Jiang et al., 2017). Although numerous studies have demonstrated differences in gene transcription among genetic groups or lines in aquatic species (Langevin et al., 2012; Marancik et al., 2014; Reyes-López et al., 2015; Robledo et al., 2016), few have partitioned genetic variation and estimated heritability for gene transcription (Roberge et al., 2007; Normandeau et al., 2009; Aykanat et al., 2012; He et al., 2017).
Gene expression variation can be considered as a phenotype (Houle et al., 2010), and as with any traditional phenotypic trait, gene expression variation is influenced by various genetic and environmental factors (Leder et al., 2015). It is clear that gene expression variation is widespread among individuals and taxa and can be heritable (Whitehead and Crawford, 2006; Dixon et al., 2007; Powell et al., 2013). Additive genetic effects of gene expression represent evolutionary potential, which allows us to predict the magnitude of its response to selective pressure (Leder et al., 2015; He et al., 2017). Nonadditive effects also contribute significantly to phenotypic variation but are not heritable (Evans and Neff, 2009). All these components of genetic parameters are important contributors to phenotypic variation. Therefore, it is fundamentally important to disentangle the contributions of additive genetic and non-additive genetic variance to overall gene expression variance. This information of genetic parameter is critical for evaluating the potential for gene-expression performance markers to be used in selection programs targeting resistant clam broodstocks.
Recent studies have shown that the basal expression levels of one set of Vibrio resistance-related genes (Big-Def, BIRC7, NFIL3, CTL9, and Bax) significantly correlated with survival in challenge tests in clam M. petechialis (Jiang et al., 2018). Therefore, it seemed that selection decisions could be made based on the expression level of resistance-related genes that are genetically correlated with disease resistance. The indirect selection by gene expression trait would simplify the data collection, avoid the challenge testing, and reducing costs and time consumption. Theoretically, higher additive genetic effects reflect higher adaptive potential in response to selection pressures; thus, the gene transcription traits closely linked to phenotype and with higher additive genetic component are better candidates for an indirect selection criterion in practice. However, the genetic variance components for the transcription of resistancerelated genes and their genetic correlations with body size have not been evaluated in M. petechialis. Therefore, in this study, an animal model was used to estimate the genetic variation component for Vibrio resistance-related genes, and estimate the genetic correlations between Vibrio resistance-related genes and body sizes. The results could provide some key parameters for understanding of the potential resistant selection response of gene transcription for resistancerelated genes in non-model organisms.
2 MATERIAL AND METHOD 2.1 Experimental familiesExperimental families were produced in a hatchery at the Zhejiang Mariculture Research Institute (Wenzhou, China). In July 2014, 11 two-year-old males and 11 two-year-old females were randomly selected from the clam population and used to produce 23 full-sibling families according to a slightly modified Berg and Henryon design (Berg and Henryon, 1999). For example, each sire was mated with 2–3 dams, and each dam was mated with 2–3 sires. Thus 11 dam halfsib families and 11 sire half-sib families were produced, the detailed breeding design for these fullsibling families was presented in Fig. 1. Detailed rearing conditions for larvae have been described by Wang et al. (2011). The full-sibling families of juvenile clams were randomly distributed in pond at the Zhejiang Mariculture Research Institute. In the farming process, different families were reared separately by mesh in the same pond and exposed to similar environmental conditions to minimize the effects of uncontrolled environmental factors. In detail, the stocking density of each family was the same, and the average survival rate of families was up to 92.4%±1.9% at 15-month-old, which means that no accidental mass mortalities occurred in the cultivation.

|
| Fig.1 The schematic diagram of clam M. petechialis 23 fullsibling families 11 sires (S1-S11) and 11 dams (D1-D11) were chosen randomly for producing 23 full-sibling families. |
Before the estimation of genetic parameters, six tissues (digestive gland, mantle, gill, foot, adductor muscle, and hemocytes) were collected from four healthy clams for analysis of the expression of the detected genes between tissues. Hemolymph (~500 μL) was withdrawn from each adult clam with a 1-mL disposable syringe, and was then centrifuged at 3 000×g for 10 min at 4℃. The pellet was used in total RNA extraction. Another five tissues from each clam were taken, dissected, immediately frozen in liquid nitrogen, and stored at -80℃ until tissue distribution analysis could be conducted.
The 12-month- and 15-month-old clams from two batches were used to estimate genetic parameters respectively. We randomly selected five individuals from each family from each sampling batch for the genetic analysis. The shell width (SW) and total weight (TW) of 12-month-old clams were measured using a digital Vernier caliper and a digital balance respectively. After measuring the SW and TW, the digestive gland from each clam was dissected, immediately frozen in liquid nitrogen, and stored at -80℃ until further processing.
2.3 RNA extraction and cDNA synthesisTotal RNA was extracted from samples using an EZNA® Total RNA Kit II (Omega Bio-Teck, USA) according to the manufacturer's instructions. RNA degradation and contamination were monitored on 1% agarose gels and then quantified using a NanoDrop-1000 spectrophotometer (Thermo Scientific, USA). cDNA was synthesized from total RNA (1 μg) using a PrimeScriptTM RT reagent kit with gDNA Eraser (TaKaRa, Japan) according to the manufacturer's protocol.
2.4 Semi-quantitative RT-PCRSemi-quantitative RT-PCR was performed to determine the expression levels of Vibrio resistancerelated genes in different tissues from adult clams. The elongation factor 1α gene (EF1α) was used as the internal reference gene in semi-quantitative RT-PCR analysis (Jiang et al., 2017). Primer sequences are listed in Table 1. PCRs were performed in a 25 μL reaction volume containing 1 μL cDNA, 0.2 mmol/L of each primer, and 12.5 μL Premix TaqTM (TaKaRa, Japan). The appropriate number of cycles of these six genes were selected, 18 cycles for EF1α, 26 cycles for Bax, NFIL3 and Big-Def and 28 cycles for BIRC7 and CTL9. These cycling parameters could ensure that the amplification product is clearly visible and can be quantified. That is the amplification is in the exponential range and has not reached a plateau yet. The cycling protocol was one cycle of 94℃ for 4 min; 18, 26 or 28 cycles of 94℃ for 15 s; 55℃ for 15 s; and 72℃ for 15 s, and the final extension time was 7 min. PCR was conducted using a Takara thermocycler. PCR products were separated via electrophoresis in 2% agarose gels that were stained with 4S Red Plus Nucleic Acid Stain (Sangon, China) and visualized under ultraviolet light. The quantification of bands was performed by Bio-Rad Quantity One.
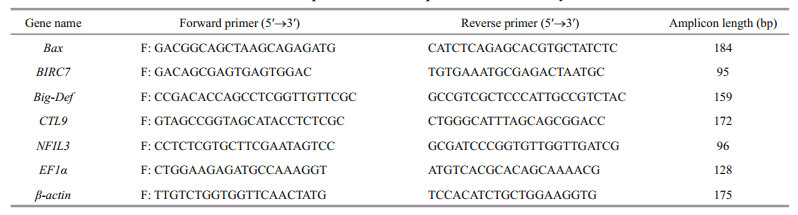
Based on a previous study (Jiang et al., 2018), 5 Vibrio resistance-related genes were selected for detection of the genetic parameters of gene expression levels using clams randomly selected from the 23 full sibling families by quantitative real-time PCR (qRTPCR). The β-actin and EF1α genes were used as internal references to normalize the relative expression levels between samples (Jiang et al., 2017). The primers for qRT-PCR are the same used for semiquantitative RT-PCR. qRT-PCR was performed in QuantStudio 6 Flex (Applied Biosystems, USA) machine using a QuantiNova SYBR Green PCR Kit (Qiagen, Germany). Each reaction was performed in triplicate under the following conditions: 95℃ for 2 min, 40 cycles of 95℃ for 5 s, and 60℃ for 20 s, followed by melting curve determination. The ΔCt value was used to quantify the relative gene expression value, the greater the ΔCt value, the lower the gene relative expression level (Pfaffl, 2001).
2.6 Estimation of genetic parametersThe phenotypic variance of 12-month- and 15-month-old clams were estimated using the following animal linear model:
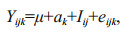
where Yijk is the phenotypic value of kth offspring of ith sire and jth dam; μ is the mean phenotype of the sample; ak is random additive genetic effect of clam k; Iij is the no additive effect due to the interaction of ith sire and jth dam; and eijk is the residual of the kth individual of ith sire and jth dam.
For the animal linear model, heritability was calculated as follows:
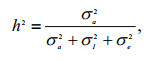
where σa2 is additive genetic variance components; σI2 interaction between sire and dam, also named family variance components; and σe2 residual variance components.
Gene transcription variance was partitioned into additive genetic variance (σa2) and the interaction between sire and dam (σI2) separately. Wilcoxon signed-rank test was conducted to compare the genetic parameters of gene transcription trait between 12-month- and 15-month-old clams.
2.7 Estimation of phenotypic and genetic correlationsThe phenotypic variance SW and TW were
partitioned using the following animal linear model:
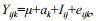
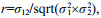
The expression levels of the five genes varied among different tissues (Fig. 2). For example, CTL9 had the highest expression level in the digestive gland, relatively low expressive abundance in hemocytes and adductor muscles, and no expression in the mantle, gill, and foot. The other four genes were expressed in all six tissues, but expression levels varied among tissues. For example, the apoptosisrelated genes Bax and BIRC7 both had the highest expression levels in the gills but were only moderately high in the digestive gland. The transcription factor of inflammatory response NFIL3 had high expression level in the mantle, gill, and foot and relatively low expression in the other three tissues. Big-Def had low expression in the gills and high expression levels in other five tissues, with the highest in the digestive gland. These results revealed that expression of all five genes could be detected in the digestive gland and hemocytes in M. petechialis. Considering that few hemocytes could be extracted from the clams, the digestive gland was selected as the major source for the following gene expression analysis.

|
| Fig.2 Expression of five Vibrio resistance-related genes mRNA in adult tissues EF1α is the internal reference gene. |
The number of observations, means, standard deviations, coefficients of variation (CV) for the five gene transcription traits (Bax, BIRC7, Big-Def, CTL9, and NFIL3) in two growth stages are shown in Table 2. The expression level differed among the five genes in digestive gland. Big-Def had the highest relative expression level and the highest CV in 12- and 15-month-old clam group, whereas the relative expression level of BIRC7 was the lowest. Furthermore, the gene expression levels of 12- and 15-month-old clam were compared. With the exception of BIRC7, other genes showed lower expression in 15-month-old clam group. The gene expression difference for five genes between 12- and 15-month-old clam group was 8%–23%. Compared to other genes, the expression of BIRC7 and NFIL3 for 12-month-old was more similar to that of 15-month-old clam group. The result of t-test suggests that expression level of the 5 genes between 12- and 15-month old clam were statistically and significantly different (P < 0.05).
|
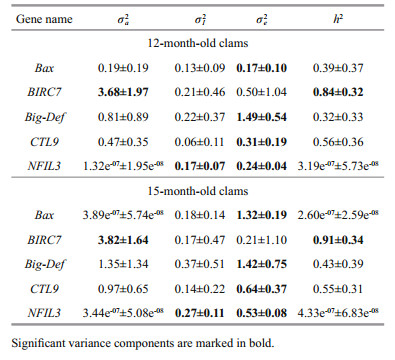
Estimation of the variance components and heritability of gene transcription trait in 12-monthand 15-month-old clams are presented in Table 3. The gene transcription trait of two different growth stages exhibited substantial similarities in genetic variance components. Across all five genes, the gene transcription traits of two stages were no significant different in additive genetic effects (Wilcoxon signedrank test, P=0.31) and sire-by-dam interaction effects (no additive genetic effects) (P=0.13). In detail, the BIRC7 gene showed significant additive genetic variations in the 12-month and 15-month-old clams, correspondingly the high heritabilities 0.84±0.32 and 0.91±0.34, respectively. Whereas, the heritabilities of other four genes (Big-Def, BAX, NFIL3, and CTL9) were low-to-moderate but not significantly. The NFIL3 gene showed significant sire-by-dam interaction effects.
|
The phenotypic and genetic correlations were analyzed based on the body size SW, TW and expression of BIRC7 gene in the stage of 12-monthold clams. The phenotypic and genetic correlation coefficients between SW and BIRC7 gene were 0.05±0.15, -0.04±0.44, the phenotypic and genetic correlation coefficients between TW and BIRC7 gene were -0.18±0.15 and -0.97±0.98 respectively. No significant phenotypic and genetic correlations were detected between BIRC7 gene transcription trait and body size in the 23 full sibling families.
4 DISCUSSIONThe basal gene expression level could vary markedly in different organs of the same animal (Engwerda and Kaye, 2000). Therefore, the choice of research organ is crucial for gene expression studies. In our experiment, all the five resistance-related genes were highly expressed in hemocytes and digestive gland in M. petechialis. It has been shown that hemocytes play a key role in animal immune response (Lavine and Strand, 2002). However, in M. petechialis, due to few hemocytes extracted from the clams' bodies, it is not a preferred organ at the individual level or for batch analyses. In mollusks, the digestive gland is another important tissue of the immune system because their epithelial cells can produce major immune molecules (Röszer, 2014). In addition, the digestive gland has been used as a preferred research organ in M. petechialis (Gao et al., 2012; Zou and Liu, 2016). In this study, the digestive gland is the preferred tissue for gene expression analysis considering that all five genes expressed in digestive gland and it could be collected conveniently.
Gene expression variation can underlie complex phenotype variations (Ayroles et al., 2009) and play important roles in the response to environmental change and stress (Crawford and Powers, 1992). It has long been thought that a large part of gene expression variation is highly heritable with an underlying polygenic architecture (Cheung and Spielman, 2002; Gilad et al., 2008). In recent years, the genetic parameter of gene transcription has been estimated in some aquatic species (Aykanat et al., 2012; Brokordt et al., 2015; Leder et al., 2015; He et al., 2017). Additionally, the heritability of different gene transcription was diverse. A potential explanation is that different phenotypic or transcription traits undergo different strength of selection throughout an animal's life. For example, as those closely related with the biological fitness, i.e. for survival and reproduction etc., undergo more strong selective pressures than other traits, and usually show lower heritabilities. Herein, we examined the genetic parameters of gene transcription for five Vibrio resistance-related genes in full sibling clam families. The results showed that the heritability of BIRC7 was 0.91 and the heritability of other four genes was not different from zero, which showed that different gene transcription traits could exhibit dramatic variation in heritability. In previous studies, Aykanat et al. (2012) found that the heritability of four cytokine genes in Chinook salmon varied from 0.05 to 0.44, and Tedeschi et al. (Tedeschi et al., 2016) reported a higher heritability of three heat shock protein genes in sea turtles (mean h2=0.58). This presence of heritability suggests that Vibrio resistance-related gene expression variation of BIRC7 gene are heritable. Thus, BIRC7 gene transcription may provide reliable genetically based markers for use in selective breeding and the performance of gene transcription can be improved by successive selection.
Genetic analysis is heavily dependent on data, a larger data set will provide a more accurate estimation of genetic parameters, but the costs used in getting the data will also be increased. No research yet has provided the evidence that appropriate sample size is required for genetic analysis. Due to the measuring procedure is complicated for transcription traits and the high cost, the sample size used in genetic analysis is usually not large. For example, Aykanat et al. (2012) used a data set including 48 families and 192 individuals to partition the transcription variation of four cytokine genes into additive genetic, nonadditive genetic, and maternal components in Chinook salmon Oncorhynchus tshawytscha. In our study, we used 23 full sibling families to estimate the genetic parameters of 12-month- and 15-month-old clams' transcription traits. The results showed that there was no significant differences in additive genetic effects and sire-by-dam interaction effects between two growth stages (Wilcoxon signed-rank test P=0.31, P=0.13), which indicated that the genetic variance components of gene expression traits were relatively stable during short-term growth. However, the estimation of heritability in our study was accompanied by high standard error, which are likely associated with low sample sizes, and possibly resulted in our failure to detect significant low-to-moderate heritabilities (Table 3). Nevertheless, we observed significant additive effects in BIRC7, which might be valuable in the genetic improvement of Vibrio resistance in artificial selection approaches.
Animals are frequently faced with trade-offs between growth and mortality rates. Enhancement of disease resistance may become with compromise in growth traits. For instance, Yáñez et al. (2016) has reported a significant negative genetic correlation between resistance against Piscirickettsia salmonis and harvest weight in coho salmon. Furthermore, a slightly negative genetic correlation between resistance to vibriosis and harvest weight was found in Atlantic cod (Bangera et al., 2011). These results suggest that selective breeding for faster growth may have a negative effect on disease resistance in these species. Conversely, a significant positive genetic correlation between resistance to fish pasteurellosis and body length was reported in the gilthead sea bream (Antonello et al., 2009), which may be due to resistant individuals experiencing superior growth. Additionally, genetic correlations that are not significantly different from zero have also been found between disease resistance and harvest weight in some aquatic species (Silverstein et al., 2009; FloresMara et al., 2017). Results of these studies suggest that genetic correlations between disease resistance and growth of different species are not uniform. Therefore, genetic correlation must be evaluated case by case in practical breeding. In this study, we found that there is no significant genetic correlation between Vibrio resistance-related gene BIRC7 and body sizes. Our results indicate that variation in gene transcription could help the genetic improvement of Vibrio resistance and will not influence body size in M. petechialis.
5 CONCLUSIONIn conclusion, our results first demonstrate the variation in the expression of the Vibrio resistancerelated BIRC7 gene in M. petechialis is heritable, indicating the feasibility of improving this trait by artificial selection. Moreover, no significant genetic correlation is detected between the Vibrio resistancerelated BIRC7 gene and body sizes, suggesting that artificial selection for Vibrio-resistance will not influence body size in M. petechialis.
6 DATA AVAILABILITY STATEMENTData sharing not applicable to this article as no datasets were generated or analyzed during the current study. All data generated and/or analyzed during this study are included in this published article.
Antonello J, Massault C, Franch R, Haley C, Pellizzari C, Bovo G, Patarnello T, De Koning D J, Bargelloni L. 2009. Estimates of heritability and genetic correlation for body length and resistance to fish pasteurellosis in the gilthead sea bream (Sparus aurata L. ). Aquaculture, 298(1-2): 29-35.
DOI:10.1016/j.aquaculture.2009.10.022 |
Aykanat T, Heath J W, Dixon B, Heath D D. 2012. Additive, non-additive and maternal effects of cytokine transcription in response to immunostimulation with Vibrio vaccine in Chinook salmon (Oncorhynchus tshawytscha). Immunogenetics, 64(9): 691-703.
DOI:10.1007/s00251-012-0624-2 |
Ayroles J F, Carbone M A, Stone E A, Jordan K W, Lyman R F, Magwire M M, Rollmann S M, Duncan L H, Lawrence F, Anholt R R, Mackay T F. 2009. Systems genetics of complex traits in Drosophila melanogaster. Nature Genetics, 41(3): 299-307.
DOI:10.1038/ng.332 |
Bangera R, Ødegård J, Præbel A K, Mortensen A, Nielsen H M. 2011. Genetic correlations between growth rate and resistance to vibriosis and viral nervous necrosis in Atlantic cod (Gadus morhua L). Aquaculture, 317(1-4): 67-73.
DOI:10.1016/j.aquaculture.2011.04.018 |
Berg P, Henryon M. 1999. Selection response under alternative mating designs in fish. Proc. Adv. Anim. Breed. Gen., 13: 297-300.
|
Bishop S C, Woolliams J A. 2014. Genomics and disease resistance studies in livestock. Livestock Science, 166: 190-198.
DOI:10.1016/j.livsci.2014.04.034 |
Brokordt K B, González R C, Farías W J, Winkler F M. 2015. Potential response to selection of HSP70 as a component of innate immunity in the abalone Haliotis rufescens. PLoS One, 10(11): e0141959.
DOI:10.1371/journal.pone.0141959 |
Cheung V G, Spielman R S. 2002. The genetics of variation in gene expression. Nature Genetics, 32(S4): 522-525.
DOI:10.1038/ng1036 |
Crawford D L, Powers D A. 1992. Evolutionary adaptation to different thermal environments via transcriptional regulation. Molecular Biology and Evolution, 9(5): 806-813.
|
Dixon A L, Liang L M, Moffatt M F, Chen W, Heath S, Wong K C C, Taylor J, Burnett E, Gut I, Farrall M, Lathrop G M, Abecasis G R, Cookson W O. 2007. A genome-wide association study of global gene expression. Nature Genetics, 39(10): 1 202-1 207.
DOI:10.1038/ng2109 |
Engwerda C R, Kaye P M. 2000. Organ-specific immune responses associated with infectious disease. Immunology Today, 21(2): 73-78.
DOI:10.1016/S0167-5699(99)01549-2 |
Evans M L, Neff B D. 2009. Non-additive genetic effects contribute to larval spinal deformity in two populations of Chinook salmon (Oncorhynchus tshawytscha). Aquaculture, 296(1-2): 169-173.
DOI:10.1016/j.aquaculture.2009.08.018 |
Flores-Mara R, Rodríguez F H, Bangera R, Lhorente J P, Neira R, Newman S, Yáñez J M. 2017. Resistance against infectious pancreatic necrosis exhibits significant genetic variation and is not genetically correlated with harvest weight in rainbow trout (Oncorhynchus mykiss). Aquaculture, 479: 155-160.
DOI:10.1016/j.aquaculture.2017.05.042 |
Gao X G, He C B, Liu H, Li H J, Zhu D, Cai S L, Xia Y, Wang Y, Yu Z. 2012. Intracellular Cu/Zn superoxide dismutase(Cu/Zn-SOD) from hard clam Meretrix meretrix:its cDNA cloning, mRNA expression and enzyme activity. Molecular Biology Reports, 39(12): 10 713-10 722.
DOI:10.1007/s11033-012-1962-8 |
Gilad Y, Rifkin S A, Pritchard J K. 2008. Revealing the architecture of gene regulation:the promise of eQTL studies. Trends in Genetics, 24(8): 408-415.
DOI:10.1016/j.tig.2008.06.001 |
Gilmour A R, Gogel B, Cullis B, Thompson R, Butler D. 2009.ASReml User Guide Release 3.0. VSN International Ltd., Hemel Hempstead, UK.
|
He X, Houde A L S, Pitcher T E, Heath D D. 2017. Genetic architecture of gene transcription in two Atlantic salmon(Salmo salar) populations. Heredity (Edinb), 119(2): 117-124.
DOI:10.1038/hdy.2017.24 |
Houle D, Govindaraju D R, Omholt S. 2010. Phenomics:the next challenge. Nature Reviews Genetics, 11(12): 855-866.
DOI:10.1038/nrg2897 |
Jiang F J, Wang H X, Yue X, Zhang S J, Liu B Z. 2018. Integrating the Vibrio-resistance phenotype and gene expression data for discovery of markers used for resistance evaluation in the clam Meretrix petechialis. Aquaculture, 482: 130-136.
DOI:10.1016/j.aquaculture.2017.09.033 |
Jiang F J, Yue X, Wang H X, Liu B Z. 2017. Transcriptome profiles of the clam Meretrix petechialis hepatopancreas in response to Vibrio infection. Fish & Shellfish Immunology, 62: 175-183.
|
Langevin C, Blanco M, Martin S A, Jouneau L, Bernardet J F, Houel A, Lunazzi A, Duchaud E, Michel C, Quillet E, Boudinot P. 2012. Transcriptional responses of resistant and susceptible fish clones to the bacterial pathogen Flavobacterium psychrophilum. PLoS One, 7(6): e39126.
DOI:10.1371/journal.pone.0039126 |
Lavine M D, Strand M R. 2002. Insect hemocytes and their role in immunity. Insect Biochemistry and Molecular Biology, 32(10): 1 295-1 309.
DOI:10.1016/S0965-1748(02)00092-9 |
Leder E H, McCairns R J, Leinonen T, Cano J M, Viitaniemi H M, Nikinmaa M, Primmer C R, Merilä J. 2015. The evolution and adaptive potential of transcriptional variation in sticklebacks——signatures of selection and widespread heritability. Molecular Biology and Evolution, 32(3): 674-689.
DOI:10.1093/molbev/msu328 |
Liang B B, Jiang F J, Zhang S J, Yue X, Wang H X, Liu B Z. 2017. Genetic variation in vibrio resistance in the clam Meretrix petechialis under the challenge of Vibrio parahaemolyticus. Aquaculture, 468: 458-463.
DOI:10.1016/j.aquaculture.2016.10.037 |
Liu B Z, Dong B, Tang B J, Zhang T, Xiang J H. 2006. Effect of stocking density on growth, settlement and survival of clam larvae, Meretrix meretrix. Aquaculture, 258(1-3): 344-349.
|
Marancik D, Gao G T, Paneru B, Ma H, Hernandez A G, Salem M, Yao J B, Palti Y, Wiens G D. 2014. Whole-body transcriptome of selectively bred, resistant-, control-, and susceptible-line rainbow trout following experimental challenge with Flavobacterium psychrophilum. Front Genetiers, 5: 453.
|
Nie Q, Yue X, Chai X L, Wang H X, Liu B Z. 2013. Three vibrio-resistance related EST-SSR markers revealed by selective genotyping in the clam Meretrix meretrix. Fish& Shellfish Immunology, 35(2): 421-428.
|
Nie Q, Yue X, Liu B Z. 2015. Development of Vibrio spp. infection resistance related SNP markers using multiplex SNaPshot genotyping method in the clam Meretrix meretrix. Fish & Shellfish Immunology, 43(2): 469-476.
|
Normandeau E, Hutchings J A, Fraser D J, Bernatchez L. 2009. Population-specific gene expression responses to hybridization between farm and wild Atlantic salmon. Evolutionary Applications, 2(4): 489-503.
DOI:10.1111/j.1752-4571.2009.00074.x |
Pfaffl M W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research, 29(9): e45.
DOI:10.1093/nar/29.9.e45 |
Powell J E, Henders A K, McRae A F, Kim J, Hemani G, Martin N G, Dermitzakis E T, Gibson G, Montgomery G W, Visscher P M. 2013. Congruence of additive and nonadditive effects on gene expression estimated from pedigree and SNP data. PLoS Genetics, 9(5): e1003502.
DOI:10.1371/journal.pgen.1003502 |
Reyes-López F E, Romeo J S, Vallejos-Vidal E, Reyes-Cerpa S, Sandino A M, Tort L, Mackenzie S, Imarai M. 2015. Differential immune gene expression profiles in susceptible and resistant full-sibling families of Atlantic salmon (Salmo salar) challenged with infectious pancreatic necrosis virus (IPNV). Developmental & Comparative Immunology, 53(1): 210-221.
|
Roberge C, Guderley H, Bernatchez L. 2007. Genomewide identification of genes under directional selection:gene transcription QST scan in diverging Atlantic salmon subpopulations. Genetics, 177(2): 1 011-1 022.
DOI:10.1534/genetics.107.073759 |
Robinson N, Hayes B. 2008. Modelling the use of gene expression profiles with selective breeding for improved disease resistance in Atlantic salmon (Salmo salar). Aquaculture, 285(1-4): 38-46.
DOI:10.1016/j.aquaculture.2008.08.016 |
Robledo D, Taggart J B, Ireland J H, McAndrew B J, Starkey W G, Haley C S, Hamilton A, Guy D R, Mota-Velasco J C, Gheyas A A, Tinch A E, Verner-Jeffreys D W, Paley R K, Rimmer G S, Tew I J, Bishop S C, Bron J E, Houston R D. 2016. Gene expression comparison of resistant and susceptible Atlantic salmon fry challenged with Infectious Pancreatic Necrosis virus reveals a marked contrast in immune response. BMC Genomics, 17: 279.
DOI:10.1186/s12864-016-2600-y |
Röszer T. 2014. The invertebrate midintestinal gland("hepatopancreas") is an evolutionary forerunner in the integration of immunity and metabolism. Cell and Tissue Research, 358(3): 685-695.
DOI:10.1007/s00441-014-1985-7 |
Silverstein J T, Vallejo R L, Palti Y, Leeds T D, Rexroad III C E, Welch T J, Wiens G D, Ducrocq V. 2009. Rainbow trout resistance to bacterial cold-water disease is moderately heritable and is not adversely correlated with growth. Journal of Animal Science, 87(3): 860-867.
DOI:10.2527/jas.2008-1157 |
Tedeschi J N, Kennington W J, Tomkins J L, Berry O, Whiting S, Meekan M G, Mitchell N J. 2016. Heritable variation in heat shock gene expression:a potential mechanism for adaptation to thermal stress in embryos of sea turtles. Proceedings Biological Sciences, 283(1822): 20152320.
DOI:10.1098/rspb.2015.2320 |
Wang H X, Chai X L, Liu B Z. 2011. Estimation of genetic parameters for growth traits in cultured clam Meretrix meretrix (Bivalvia:Veneridae) using the Bayesian method based on Gibbs sampling. Aquaculture Research, 42(2): 240-247.
DOI:10.1111/j.1365-2109.2010.02617.x |
Whitehead A, Crawford D L. 2006. Variation within and among species in gene expression:raw material for evolution. Molecular Ecology, 15(5): 1 197-1 211.
DOI:10.1111/j.1365-294X.2006.02868.x |
Yáñez J M, Bangera R, Lhorente J P, Barría A, Oyarzún M, Neira R, Newman S. 2016. Negative genetic correlation between resistance against Piscirickettsia salmonis and harvest weight in coho salmon (Oncorhynchus kisutch). Aquaculture, 459: 8-13.
DOI:10.1016/j.aquaculture.2016.03.020 |
Yang J, Benyamin B, McEvoy B P, Gordon S, Henders A K, Nyholt D R, Madden P A, Heath A C, Martin N G, Montgomery G W, Goddard M E, Visscher P M. 2010. Common SNPs explain a large proportion of the heritability for human height. Nature Genetics, 42(7): 565-569.
DOI:10.1038/ng.608 |
Zou L H, Liu B Z. 2016. The polymorphisms of a MIF gene and their association with Vibrio resistance in the clam Meretrix meretrix. Developmental & Comparative Immunology, 62: 116-126.
|