 2021, Vol. 39
2021, Vol. 39Institute of Oceanology, Chinese Academy of Sciences
Article Information
- SONG Xiaoyue, ZENG Jiangning, ZHOU Yi, CHEN Quanzhen, YANG Hongsheng, SHOU Lu, LIAO Yibo, HUANG Wei, DU Ping, LIU Qiang
- Partial function prediction of sulfate-reducing bacterial community from the rhizospheres of two typical coastal wetland plants in China
- Journal of Oceanology and Limnology, 39(1): 185-197
- http://dx.doi.org/10.1007/s00343-019-9177-7
Article History
- Received Jul. 12, 2019
- accepted in principle Sep. 16, 2019
- accepted for publication Dec. 29, 2019




2 CAS Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China;
3 Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China
Coastal wetlands are among the most significant ecosystems around the world and provide many crucial ecological services. The rhizosphere microbes of aquatic plants, which function to support the nutrition, health, and quality of the plants as a dynamic and complex interface for chemical, physical, and biological interactions, play important roles in the biogeochemical processes in coastal wetlands (Bais et al., 2006; Berg and Smalla, 2009). It has been shown that the activities of sediment bacteria are coupled to the photosynthetic activity of the plants, through the excretion of dissolved organic compounds into the rhizosphere (Welsh et al., 1997; Cifuentes et al., 2003). The abundant vegetation-derived organic substances deposited in coastal wetlands are typically mineralized through anaerobic processes— predominantly methanogenesis, and especially, sulfate reduction. Previous studies have shown that sulfate reduction under non-limiting sulfate conditions executed by sulfate-reducing bacteria (SRB), accounts for up to 50% of carbon mineralization in marine sediments (Jørgensen, 1982; Fauque, 1995; Rabus et al., 2006). SRB, which are ubiquitous anaerobic microorganisms that primarily use sulfate as their terminal electron acceptor to complete the decomposition of organic matter, play significant roles in the global biogeochemical cycles (Steudler and Peterson, 1984; Muyzer and Stams, 2008). Dissimilatory sulfate reduction has evolved for 3.5 billion years, as indicated by the stable sulfur isotopes; thus, several SRB are regarded as ancestral microorganisms of this planet (Shen and Buick, 2004; Habicht et al., 2005; Barton and Fauque, 2009). SRB have successfully adapted to almost all ecosystems on the Earth, especially in some extreme environments, such as deep-sea hydrothermal vents and oil fields. Coastal wetlands are one of the major habitats of SRB, and since these microbes depend on sulfate for their metabolic activity, marine wetland sediments contain a larger proportion of SRB (up to 20%–40% of all bacteria) than freshwater wetlands do (Pester et al., 2012; Cui et al., 2017). Furthermore, SRB exhibit high sulfate-reducing activity and play significant roles in organic carbon remineralization, benthic geochemistry, and plant-microbe interactions (Hines et al., 1989; Howarth, 1993; Rooney-Varga et al., 1997; Hines et al., 1999). In particular, high sulfate reduction rates have been measured in the rhizospheres of numerous marine macrophytes, such as the eelgrass species Zostera marina and Zostera noltii and the small cordgrass Spartina maritima (Blaabjerg et al., 1998; Nielsen et al., 2001).
The dissimilatory sulfate reduction pathway from sulfate to sulfite involves three enzymes: sulfate adenylyltransferase (encoded by the sat gene), adenylylsulfate reductase (encoded by the aprAB gene), and dissimilatory-type sulfite reductase (encoded by the dsrAB gene). The calculation of the proportion of dissimilatory sulfate reduction genes is based on the total number of genes encoding these enzymes in each sample. Currently, SRB are thought to be composed of two groups, namely, those that degrade organic compounds completely to carbon dioxide, and those that degrade organic compounds incompletely to acetate (Muyzer and Stams, 2008). Some genera of SRB species, such as Desulfovibrio and Desulfotomaculum, utilize hydrogen and incompletely oxidize several organic compounds (e.g., ethanol, formate, lactate, pyruvate, malate, and succinate) to acetate for growth. Other genera, including Desulfobacter and Desulfobacterium, are able to decompose organic substrates completely to carbon dioxide by further oxidizing the acetate. Because the process of acetate conversion to acetylCoA is essential for complete degradation, the acetylCoA synthetase gene proportion has been adopted to indicate the ability of a sulfate-reducing bacterial community to completely degrade organic substrates.
To obtain additional knowledge of the biogeochemical and ecological roles of SRB in coastal wetlands in particular, it is essential to analyze their relevant community structure and metabolic characteristics. Previous studies have shown that Desulfovibrionaceae and Desulfobacteraceae were dominant groups of SRB in the sediments of several coastal wetlands (Rooney-Varga et al., 1997; Bahr et al., 2005; Moreau et al., 2010; Zeleke et al., 2013). However, the SRB were highly variable among different areas and plant growth stages (RooneyVarga et al., 1997; Klepac-Ceraj et al., 2004; Zeleke et al., 2013). During the last few decades, the development of new molecular technologies has vastly advanced our understanding of the diversity and phylogenies of the sulfate-reducing bacterial strains existing in coastal wetlands. In the early period, 16S rRNA target probes were commonly adopted to determine particular sulfate-reducing bacterial groups (Wagner et al., 1993; Devereux and Mundfrom, 1994; Lücker et al., 2007); however, later, the high-throughput sequencing of 16S rRNA and functional genes, such as aprAB and dsrAB, became increasingly popular in studies on SRB in coastal environments (Friedrich, 2002; Guan et al., 2013; Cui et al., 2017). However, the progress in metabolic studies on SRB, which were usually focused on the utilization of substrates and limited by the efficiency of the cultivation experiments, could not be compared to that of the phylogenetic studies. Thus, there only few studies have provided sufficient information on the in-situ composition and metabolism of sulfatereducing bacterial communities simultaneously.
Previous studies have suggested that the distribution and dynamics of the community composition of SRB in coastal wetlands have close association with environmental factors, such as salinity, temperature, pH, and vegetation (Galand et al., 2002; Baldwin et al., 2006; Cadillo-Quiroz et al., 2006; Smith et al., 2008; Kotiaho et al., 2010; She et al., 2016). Nevertheless, recent studies have provided credible evidence indicating that the functional composition of microbes responds more closely to the environment factors than their taxonomic composition does (Louca et al., 2016; Gibbons et al., 2017). Thus, the elucidation of the functional profiles of microbial communities would be of great significance toward characterizing their metabolic features to a certain extent. Although metagenomic technologies are the usual approaches used to reveal the microbial community functions, the high cost and extremely complex data analysis involved limit their wide application. Based on the principle that community functions are determined by the community structure, an approach combining the 16S rDNA information of the studied sample with online-sourced 16S rDNA and microbial wholegenomic data was developed to predict the functions of the samples tested (Langille et al., 2013).
Currently, third-generation sequencing technologies are enabling the attainment of 16S rDNA information in one read (Mosher et al., 2013), thereby vastly increasing the efficiency and accuracy of the 16S rDNA sequencing of environmental samples. Therefore, in this study, microbial community function prediction based on 16S rDNA nanopore sequencing was adopted to partially determine the sulfate-reducing and organic substrate-decomposing characteristics of SRB in the rhizospheres of two typical coastal wetland plants in North and South China: Zostera japonica and Scirpus mariqueter. The proportions of genes involved in dissimilatory sulfate reduction, (i.e., sat, aprAB, and dsrAB) were used to represent the relative functional activity of this process. Additionally, the total organic carbon (TOC), total nitrogen (TN), total sulfur (TS), and water-soluble sulfate (SO42ˉ) contents in the sediments in which the two plants were planted were determined, and the phylogenetic structure, community composition, and diversity of SRB in the two plant rhizospheres were analyzed. The primary aim of the study was to test the feasibility of applying microbial community function prediction for research on the metabolic features of SRB from coastal wetlands.
2 MATERIAL AND METHOD 2.1 Site description and sediment samplingOur samples were collected from the seagrass meadows (37°21′24″N, 122°35′21″E) of Swan Lake, Rongcheng, Shandong Province, and the saltmarshes (30°22′28″N, 121°11′11″E) of Cixi, a coastal city in Zhejiang Province of China (Fig. 1). Swan Lake is famous for its flush Z. marina meadows and the whooper swans that feed on the eelgrass in winters; however, there are also stable Z. japonica meadows along the intertidal zones of this lake (Zhang et al., 2015). The Cixi saltmarshes sit along the shores of Cixi city and typically consist of zones of Phragmites australis, Spartina alterniflora, and S. mariqueter communities (Yuan et al., 2017). The obvious common ground for the Z. japonica meadows and S. mariqueter belts is that both are distributed in the lower edge of the intertidal zones and experience daily submersions under water and exposures to air.

|
| Fig.1 Zostera japonica meadows in Swan Lake (37°21′24″N, 122°35′21″E) (a); Scirpus mariqueter-comprising saltmarshes in Cixi (30°22′28″N, 121°11′11″E) (b) |
In this study, four sediment cores from the Z. japonica and S. mariqueter rhizospheres (diameter: 4.7 cm) were collected in a depth of 0–10 cm from four sites (in approximately 20 m apart from each other) in the two plant zones. The sampling sites were named using a combination of the abbreviation of the area names (CX and SL) and the order of sampling. (Hereinafter, the rhizosphere sediment samples from the S. mariqueter-comprising saltmarshes of Cixi and the Z. japonica meadows of Swan Lake are referred to simply as the CX and SL sediments, respectively.) The sampling of both areas was carried out in the winter of 2018 in an air temperature of 8–10 ℃. The sediment cores of 3–7 cm were sliced and mixed, and portions of the samples were then subjected to microbial and physicochemical analyses. The remaining samples were stored in -80 ℃ in the laboratory.
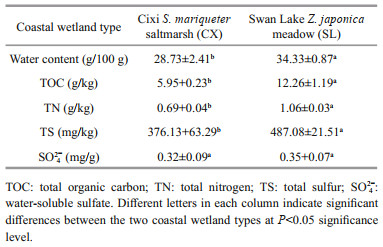
2.2 Analysis of organic matter in the sedimentsThe fresh sediment samples were lyophilized in -50 ℃ for 60 h to determine their water contents. The lyophilized samples were sieved through a 2-mm mesh before organic matter analysis. The sample used for TOC analysis was first acidized with excessive 1 mol/L HCl to remove carbonates, and the TOC content was then determined in an elemental analyzer (PE2400 Series Ⅱ, PerkinElmer, Norwalk, CT, USA). The TN and TS contents were determined using the same elemental analyzer. Water-soluble SO42ˉ was extracted from the fresh wetland sediments in a water꞉soil ratio of 2.5꞉1, and the concentrations based on the turbidity of the extracts after the addition of excessive BaCl2 were determined at 440 nm using a spectrometer. Differences in the TOC, TN, TS, and SO42ˉ levels between the Cixi and Swan Lake samples were analyzed with the Student t-test (SPSS 19.0).
2.3 Extraction, sequencing, and quantification of 16S rDNA from the sedimentsThe total genomic DNA in 0.25 g (wet weight) of each sediment sample was extracted in tubes (in duplicate) using the PowerSoil DNA Kit (Mo Bio Laboratories, Carlsbad, CA, USA) according to the manufacturer's instructions (Zeleke et al., 2013). The bacterial 16S rRNA gene was amplified with the polymerase chain reaction (PCR), using the 27F and 1492R primers (which contained a set of 16-nucleotide barcodes), for barcoded single molecule real-time (SMRT) sequencing of the full-length gene (Cao et al., 2017). The PCR program was 95 ℃ for 4 min; 30 cycles of 95 ℃ for 60 s, 60 ℃ for 45 s, and 72 ℃ for 60 s; and a final extension of 72 ℃ for 7 min (2720 Thermal Cycler, Applied Biosystems, Foster City, CA, USA) (Liu et al., 2015; Cao et al., 2017). The amplicons were sequenced using nanopore sequencing technology on a PacBio RS Ⅱ instrument (Pacific Biosciences, Menlo Park, CA, USA). The quality control for the PCR amplification and sequence preprocessing was performed as described previously (Mosher et al., 2013; Cao et al., 2017). The 16S rDNA was then subjected to SYBR Green I-based quantitative PCR (qPCR), using the primers 338f (ACTCCTACGGGAGGCAGC) and 536r (GTATTACCGCGGCKGCTG) (Zeleke et al., 2013), to determine the number of copies in each sample.
2.4 Operational taxonomic unit, phylogenetic, community composition, and diversity analyses 2.4.1 Operational taxonomic unit determinationThe raw data were processed using the RS_ ReadsOfinsert.1 protocol available on the SMRT Portal (Version 2.7, Pacific Biosciences), with default filtering parameters (Hou et al., 2015; Cao et al., 2017). The Quantitative Insights into Microbial Ecology (QIIME) package (Version 1.8.0) was applied to extract high-quality sequences (Caporaso et al., 2010), and then UCLUST (Edgar, 2010; Cao et al., 2017) was applied to align those sequences to obtain representative ones. The unique sequence set was classified into operational taxonomic units (OTUs) under the 99% threshold identity, using UCLUST (Kim et al., 2014; Cao et al., 2017). After removing the potential chimeric sequences in the representative set of OTUs with Chimera Slayer (Haas et al., 2011; Cao et al., 2017), the taxonomy of each representative OTU sequence was assigned using the SILVA database. On the basis of the published taxonomic information of SRB (Itoh et al., 1998, 1999; Castro et al., 2000; Mori et al., 2003; Rabus et al., 2006; Ollivier et al., 2007; Muyzer and Stams, 2008), the OTU sequences belonging to SRB were selected from the entire list of sediment 16S rDNA data of each sample, with their numbers in each sample. Several of the OTU sequences that were not assigned to a specific genus were submitted to a search on the National Center for Biotechnology Information (NCBI)'s Basic Local Alignment Search Tool (BLAST), at the similarity threshold of 93%, for the determination of their genus association.
2.4.2 Phylogenetic analysisThe OTU sequences from the CX and SL samples were respectively edited and aligned manually, using Geneious Prime with default settings. Notably, only the OTUs whose proportion surpassed 1% of the total OTU numbers of SRB were used to build the phylogenetic trees of these bacteria in the two study areas. The reference sequences were chosen from the NCBI database, which showed the closest genetic relationships with OTUs selected through the online BLAST search. The phylogenetic relationships were estimated using the Bayesian inference and maximumlikelihood methods. FigTree (Version 1.4.3) (Rambaut, 2016) was used to visualize the tree files. The maximum-likelihood analysis was performed with raxmlGUI (Version 1.5b2) (Silvestro and Michalak, 2012), with a selection of ML+rapid bootstrap and support assessment using 1 000 rapid bootstraps.
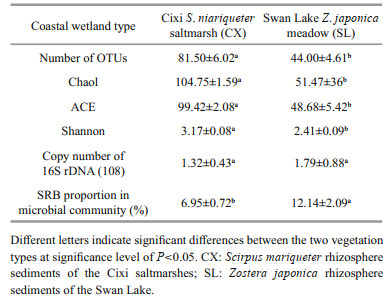
2.4.3 Community composition and diversity analysesOn the basis of the taxonomic assignments, the number of OTUs in specific families were calculated, and the community compositions of SRB in the two study areas were calculated in term of the proportions of these families and the Order. Before calculating the diversity indices, the sequence numbers of each sample were normalized to an equal number (Zeleke et al., 2013). The calculation of three species richness and diversity indices (Chao1, ACE, and Shannon) was performed using Mothur (Version 1.30). The differences in diversity indices and OTU numbers between the CX and SL samples were analyzed with the Student t-test (SPSS 19.0), respectively.
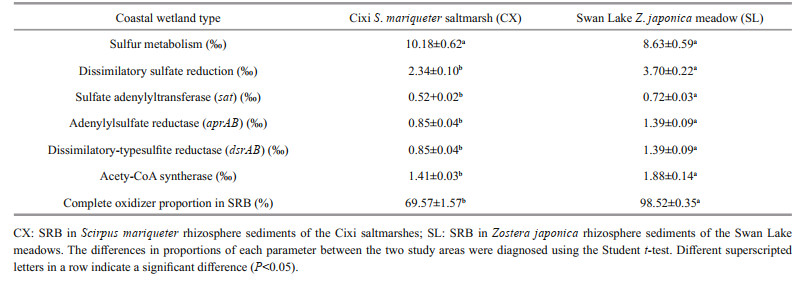
2.5 Partial function prediction of SRBThe sulfur metabolism, dissimilatory sulfate reduction, and acetyl-CoA synthesis functions of the SRB were predicted in terms of the corresponding gene proportions among all functional genes. The bioinformatics software package PICRUSt was applied to obtain information about the Kyoto Encyclopedia of Genes and Genomes (KEGG) families of every OTU sequence of SRB. By combing through the table of normalized total sequence numbers of OTUs, a table of the KEGG Orthology (KO) counts of all the functional genes in each sample was acquired. The pathway description and Enzyme Commission (EC) number of every KO identifier were searched from the KEGG databases. By classifying and summarizing the potential functional genes of SRB in each sample, the proportion of genes for sulfur metabolism was calculated, which involved all genes in the sulfur metabolism pathways (No. 00920 in KEGG) of SRB. Thereafter, the KO records of enzymes involved in dissimilatory sulfate reduction (including sulfate adenylyltransferase, adenylylsulfate reductases, and dissimilatory-type sulfite reductases) and that of acetyl-CoA synthetase were selected from that table through the sulfur and carbon metabolism diagrams in KEGG. The proportions of genes for sulfur metabolism, dissimilatory sulfate reduction, and acetyl-CoA synthetases were calculated on the basis of the table of KO counts. The differences in these proportions between the CX and SL samples were analyzed with the Student t-test (SPSS 19.0).
3 RESULT 3.1 Physicochemical characteristics of the sedimentsSignificant differences in several physicochemical parameters were observed between the CX and SL sediments (Table 1). The TOC and TN contents of the SL sediments were statistically significantly higher than those of the CX sediments (P < 0.01). In total, the organic richness of the SL sediments was also higher. There was no statistical difference in the SO42ˉ contents of the rhizosphere sediments between the two coastal wetlands. However, the TS content was remarkably higher in the SL sediments (P < 0.01).
The number of sulfate-reducing bacterial OTUs in the samples from Cixi significantly exceeded that in the sediments from Swan Lake (Table 2, P < 0.01). The species richness indices (viz., Chao1 and ACE) showed similar associations with the number of sulfate-reducing bacterial OTUs between the S. mariqueter and Z. japonica rhizospheres (P < 0.001). However, the Shannon diversity index was remarkably higher for the CX samples (P < 0.01). These results indicate that the S. mariqueter rhizosphere sediments in Cixi possessed a significantly higher diversity of SRB than the Z. japonica rhizosphere sediments in Swan Lake. The qPCR results suggest that there was no significant difference in the amount of prokaryotic microbes between the rhizosphere sediments of the two wetlands (Table 2). On the basis of the OTU counts, the proportion of SRB in the microbial community of every sample was calculated (Table 2), with the results showing that the proportion was remarkably higher in the SL sediments (P < 0.001).
|
Two phylogenetic trees (Figs. 2 & 3) were constructed based on the 16S rDNA OTU sequences from SRB in the CX and SL sediments, respectively. Only two classes, Deltaproteobacteria and Nitrospira, were found in the samples. The proportions of sequences belonging to the Deltaproteobacteria were in the range of 86.76%–90.07% in the CX samples and 98.83%–100% in the SL samples, which indicated the dominance of SRB in these two areas.

|
| Fig.2 Maximum-likelihood phylogenetic tree based on 16S rDNA OTU sequences of SRB from Scirpus mariqueter rhizosphere sediments of the Cixi saltmarshes (CX) The OTUs were defined at 99% threshold identity. The trees only contained OTUs whose proportion surpassed 1% of the total sulfate-reducing bacterial OTU number. |

|
| Fig.3 Maximum-likelihood phylogenetic trees of 16S rDNA OTU sequences of SRB from Zostera japonica rhizosphere sediments of the Swan Lake meadows (SL) The OTUs were defined at 99% threshold identity. The trees only contained OTUs whose proportion surpassed 1% of the total sulfate-reducing bacterial OTU number. |
The compositions of SRB in the two study areas are shown in Figs. 4 & 5. At the family level of typical taxonomic groups of SRB (Fig. 4), the largest Deltaproteobacteria families in the CX sediments were Desulfobulbaceae (25.53%–33.57%), Desulfarculaceae (21.04%–27.13%), and Desulfuromonadaceae (17.03%–23.06%). In contrast, Desulfarculaceae dominated the sulfate-reducing bacterial community in most of the SL samples (84.31%–92.34%), except for a single sample (SL4) whose dominant family was Desulfobulbaceae (77.05%). Desulfomicrobiaceae was present in the CX samples only (0.71%–5.01%). At the genus level (Fig. 5), the three major genera in the CX sediments were Desulfuromonas (15.73%–22.06%), Desulfosarcina (11.53%–13.83%), and Desulfopila (7.98%–13.95%). In the SL sediments, the dominant genus in SL1–SL3 was Desulfatiglans (76.64%– 85.49%), and the genera in SL4 were Desulfobulbus (44.52%) and Desulfobacterium catecholicum_group (23.97%).

|
| Fig.4 Proportions of sulfate-reducing bacterial groups based on 16S rDNA phylotypes detected in each sample from two study areas CX: SRB in Scirpus mariqueter rhizosphere sediments of the Cixi saltmarshes; SL: SRB in Zostera japonica rhizosphere sediments of the Swan Lake meadows. |

|
| Fig.5 Proportions of the three largest genera of SRB detected in each sample from two study areas CX: SRB in Scirpus mariqueter rhizosphere sediments of the Cixi saltmarshes; SL: SRB in Zostera japonica rhizosphere sediments of the Swan Lake meadows. |
The results of function prediction of SRB, in terms of the proportions of genes related to sulfur metabolism and dissimilatory sulfate reduction (i.e., sat, aprAB, and dsrAB) and of acetyl-CoA synthetase, are presented in Table 3. There was no statistically significant difference in the proportion of genes for sulfur metabolism between the S. mariqueter and Z. japonica rhizospheres (P > 0.05). However, the proportions of genes involved in dissimilatory sulfate reduction (sat, aprAB, and drsAB) were all significantly higher in the SL sediments (P < 0.01). Furthermore, the proportion of the acetyl-CoA synthetase gene was also remarkably higher in the SL samples (P < 0.01). Additionally, the proportions of complete oxidizers in the sulfate-reducing bacterial community, which were obtained on the basis of the corresponding KO characteristics, were much larger in the SL sediments (P < 0.01). These results suggest that the sulfate-reducing bacterial community in the Z. japonica seagrass meadow possessed a significantly stronger ability to degrade organic substrates completely.
|
In this study, we analyzed the features of the sulfate-reducing bacterial communities from the rhizospheres of two coastal wetland plants: S. mariqueter in the saltmarshes of Cixi and Z. japonica in the seagrass meadows of Swan Lake, where the organic nutritional status of sediments in the former was significantly higher than that in the latter. The distinct phylogenetic structure, taxonomic composition, and diversity of the sulfate-reducing bacterial communities in the two areas were revealed. Furthermore, it was revealed that the sulfate-reducing bacterial community from the Z. japonica rhizosphere had higher proportions of genes for dissimilatory sulfate reduction and acetyl-CoA synthesis, thus giving them an advantage over the SRB in the S. mariqueter rhizosphere, since these genes are essential for oxidizing acetate and degrading organic compounds completely.
The higher dissimilatory sulfate reduction capability in the Z. japonica rhizosphere seemed to correspond to its larger composition of SRB. The higher TS content in the SL sediment, on the background of a similar content of SO42ˉ substrate between the two wetland areas (Table 1), indicated that more sulfate was reduced to sulfite and sulfide in Swan Lake. In this study, the higher nutritional status in the SL sediments triggered more active sulfate reduction, which was also implied by other researchers who had studied coastal vegetation habitats (Zeleke et al., 2013; Cui et al., 2017). However, this process could be realized by more prokaryotic organisms or a larger percentage of SRB. Although the numbers of prokaryotic organisms in the two wetlands were similar, the proportion of SRB in the SL sediment was nearly twice that in the CX sediment (Table 2). Meanwhile, the larger proportion of functional genes of SRB in the SL sample, which was apparent for each dominant enzyme, contributed to its dissimilatory sulfate reduction advantage. In addition, the third factor contributing to the higher dissimilatory sulfate reduction activity in the Z. japonica rhizosphere might be the stronger ability of its sulfate-reducing bacterial community to degrade the organic substrates completely. Interestingly, these patterns for the SL sediments correlated with their remarkably lower sulfate-reducing bacterial diversity in the microbial community compared with that of the CX samples. In contrast, there was no significant difference between the two study areas in terms of the total proportion of genes for sulfur metabolism. Nevertheless, more investigations and experiments should be conducted to clarify the detailed relationship between the functional gene composition of SRB and their dissimilatory sulfate reduction capability in the Z. japonica meadows and S. mariqueter-comprising saltmarshes in China. From the results on the sulfatereducing bacterial composition, it could be suggested that the dissimilatory sulfate reduction and complete organic substrate degradation capabilities of the Desulfatiglans-dominating SRB in the SL sediments were superior to those of the SRB species in the CX sediments.
The aim of our study was to preliminarily confirm the feasibility of applying function prediction of the dissimilatory sulfate reduction activity of sulfatereducing bacterial communities in coastal wetlands, on the basis of 16S rDNA whole-length sequencing. Another merit of this study was the selection of several critical enzymes in sulfate-reducing bacterial metabolism, especially acetyl-CoA synthetase for the complete degradation of organic substrates. The utilization of nanopore sequencing vastly increased the efficiency and accuracy of our study, and the online databases of microbial taxonomy and function hugely expanded our vision in potential information mining. However, this study had one limitation, as there were gaps between the proportion of functional genes predicted by the 16S rDNA sequences and the actual relevant function of the sulfate-reducing bacterial community. This might be overcome by the progress in metagenomic research on the SRB.
This study was intended to provide new information of the community composition and functions of SRB in coastal wetlands. One of the larger environmental impacts of SRB is the production of high levels of H2S, which is a direct result of dissimilatory sulfate reduction and closely affected by organic substrate degradation. At the same time, SRB are known to decrease the emission of methane by competing with the methanogens and suppressing methanogenesis. It is commonly known that wetlands are the largest sources of natural methane, and thus the dynamics of the SRB should be a key factor for restraining greenhouse gas emissions from such areas (Muyzer and Stams, 2008; Poffenbarger et al., 2011; Bridgham et al., 2013). At present, studies on SRB are enriching our understanding of their functional features and environmental roles; for example, a multicellular filamentous species of Desulfobulbaceae, named "cable bacteria, " can mediate electric currents over a few centimeters in sediments (Nielsen et al., 2010; Schauer et al., 2014; Larsen et al., 2015; SuluGambari et al., 2016) and electrically couple the oxidation of sulfide in anoxic layers with the reduction of oxygen in the sediment. Thus, there will be a wide space for the application of community function prediction in studies on SRB based on highly efficient 16S rDNA sequencing, especially in the coastal wetlands with their complicated biogeochemical background.
5 CONCLUSIONIt is concluded that function prediction based on 16S rDNA SMRT sequencing could reveal the activity of major functional genes involved in dissimilatory sulfate reduction in a sulfate-reducing bacterial community, which shall be helpful for further understanding of the biogeochemical and ecological roles of SRB in coastal wetlands. The higher dissimilatory sulfate reduction capability of the Z. japonica meadows was derived from the larger proportion of SRB in the sediment microbial community, larger proportions of functional genes involved in dissimilatory sulfate reduction, and the stronger ability of the sulfate-reducing bacterial community to completely degrade organic substrates. However, further studies combining prediction in more detail of the sulfate-reducing function and deeper investigation on the physicochemical characteristics of the sediments are needed.
6 DATA AVAILABILITY STATEMENTThe SMRT sequences of the full-length 16S rDNA from the sediments of the coastal wetlands studied are available online (SRA accession: PRJNA54821).
7 ACKNOWLEDGMENTWe are indebted to GU Ruiting, ZHANG Xiaomei, XU Shaochun, and YUE Shidong for their cooperation in collecting samples. We thank the boatman WANG Hourong in Weihai (Shandong) for his professional service in the investigation of Swan Lake, and an anonymous private car driver in Cixi who offered generous help in taking us back to the bus station when we lost our way in the Cixi saltmarshes.
Bahr M, Crump B C, Klepac-Ceraj V, Teske A, Sogin M L, Hobbie J E. 2005. Molecular characterization of sulfatereducing bacteria in a New England salt marsh. Environmental Microbiology, 7(8): l 175-l 185.
DOI:10.1111/j.1462-2920.2005.00796.x |
Bais H P, Weir T L, Perry L G, Gilroy S, Vivanco J M. 2006. The role of root exudates in rhizosphere interactions with plants and other organisms. Annual Review of Plant Biology, 57: 233-266.
DOI:10.1146/annurev.arplant.57.032905.105159 |
Baldwin D S, Rees G N, Mitchell A M, Watson G, Williams J. 2006. The short-term effects of salinization on anaerobic nutrient cycling and microbial community structure in sediment from a freshwater wetland. Wetlands, 26(2): 455-464.
DOI:10.1672/0277-5212(2006)26[455:TSEOSO]2.0.CO;2 |
Barton L L, Fauque G D. 2009. Biochemistry, physiology and biotechnology of sulfate-reducing bacteria. Advances in Applied Microbiology, 68: 41-98.
DOI:10.1016/S0065-2164(09)01202-7 |
Berg G, Smalla K. 2009. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiology Ecology, 68(1): 1-13.
DOI:10.1111/j.1574-6941.2009.00654.x |
Blaabjerg V, Mouritsen K N, Finster K. 1998. Diel cycles of sulphate reduction rates in sediments of a Zostera marina bed (Denmark). Aquatic Microbial Ecology, 15(1): 97-102.
DOI:10.3354/ame015097 |
Bridgham S D, Cadillo-Quiroz H, Keller J K, Zhuang Q L. 2013. Methane emissions from wetlands:biogeochemical, microbial, and modeling perspectives from local to global scales. Global Change Biology, 19(5): 1 325-1 346.
DOI:10.1111/gcb.12131 |
Cadillo-Quiroz H, Brauer S, Yashiro E, Sun C, Yavitt J, Zinder S. 2006. Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central New York State, USA. Environmental Microbiology, 8(8): 1 428-1 440.
DOI:10.1111/j.1462-2920.2006.01036.x |
Cao J L, Yang J X, Hou Q C, Xu H Y, Zheng Y, Zhang H P, Zhang L B. 2017. Assessment of bacterial profiles in aged, home-made Sichuan Paocai brine with varying titratable acidity by PacBio SMRT sequencing technology. Food Control, 78: 14-23.
DOI:10.1016/j.foodcont.2017.02.006 |
Caporaso J G, Kuczynski J, Stombaugh J, Bittinger K, Bushman F D, Costello E K, Fierer N, Peña A G, Goodrich J K, Gordon J I, Huttley G A, Kelley S T, Knights D, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QⅡME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5): 335-336.
DOI:10.1038/nmeth.f.303 |
Castro H F, Williams N H, Ogram A. 2000. Phylogeny of sulfate-reducing bacteria. FEMS Microbiology Ecology, 31(1): 1-9.
DOI:10.1111/j.1574-6941.2000.tb00665.x |
Cifuentes A, Antón J, de Wit R, Rodriguez-Valera F. 2003. Diversity of bacteria and Archaea in sulphate-reducing enrichment cultures inoculated from serial dilution of Zostera noltii rhizosphere samples. Environmental Microbiology, 5(9): 754-764.
DOI:10.1046/j.1470-2920.2003.00470.x |
Cui J, Chen X P, Nie M, Fang S B, Tang B P, Quan Z X, Li B, Fang C M. 2017. Effects of Spartina alterniflora invasion on the abundance, diversity, and community structure of sulfate reducing bacteria along a successional gradient of coastal salt marshes in China. Wetlands, 37(2): 221-232.
DOI:10.1007/s13157-016-0860-6 |
Devereux R, Mundfrom G W. 1994. A phylogenetic tree of 16S rRNA sequences from sulfate-reducing bacteria in a sandy marine sediment. Applied and Environmental Microbiology, 60(9): 3 437-3 439.
DOI:10.1128/AEM.60.9.3437-3439.1994 |
Edgar R C. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19): 2 460-2 461.
DOI:10.1093/bioinformatics/btq461 |
Fauque G D. 1995. Ecology of sulfate-reducing bacteria. In: Sulfate-Reducing Bacteria. Springer, Boston, MA. p.217-241.
|
Friedrich M W. 2002. Phylogenetic analysis reveals multiple lateral transfers of adenosine-5'-phosphosulfate reductase genes among sulfate-reducing microorganisms. Journal of Bacteriology, 184(1): 278-289.
DOI:10.1128/JB.184.1.278-289.2002 |
Galand P E, Saarnio S, Fritze H, Yrjälä K. 2002. Depth related diversity of methanogen Archaea in Finnish oligotrophic fen. FEMS Microbiology Ecology, 42(3): 441-449.
DOI:10.1111/j.1574-6941.2002.tb01033.x |
Gibbons S M, Lekberg Y, Mummey D L, Sangwan N, Ramsey P W, Gilbert J A. 2017. Invasive plants rapidly reshape soil properties in a grassland ecosystem. mSystems, 2(7): e00178-16.
DOI:10.1128/mSystems.00178-16 |
Guan J, Xia L P, Wang L Y, Liu J F, Gu J D, Mu B Z. 2013. Diversity and distribution of sulfate-reducing bacteria in four petroleum reservoirs detected by using 16S rRNA and dsrAB genes. International Biodeterioration & Biodegradation, 76: 58-66.
DOI:10.1016/j.ibiod.2012.06.021 |
Haas B J, Gevers D, Earl A M, Feldgarden M, Ward D V, Giannoukos G, Ciulla D, Tabbaa D, Highlander S K, Sodergren E, Methe B, DeSantis T Z, Petrosino J F, Knight R, Birren B W. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research, 21(3): 494-504.
DOI:10.1101/gr.112730.110 |
Habicht K S, Salling L, Thamdrup B, Canfield D E. 2005. Effect of low sulfate concentrations on lactate oxidation and isotope fractionation during sulfate reduction by Archaeoglobus fulgidus Strain Z. Applied and Environmental Microbiology, 71(7): 3 770-3 777.
DOI:10.1128/AEM.71.7.3770-3777.2005 |
Hines M E, Evans R S, Sharak Genthner B R, Willis S G, Friedman S, Rooney-Varga J N, Devereux R. 1999. Molecular phylogenetic and biogeochemical studies of sulfate-reducing bacteria in the rhizosphere of Spartina alterniflora. Applied and Environmental Microbiology, 65(5): 2 209-2 216.
DOI:10.1128/AEM.65.5.2209-2216.1999 |
Hines M E, Knollmeyer S L, Tugel J B. 1989. Sulfate reduction and other sedimentary biogeochemistry in a northern New England salt marsh. Limnology and Oceanography, 34(3): 578-590.
DOI:10.4319/lo.1989.34.3.0578 |
Hou Q C, Xu H Y, Zheng Y, Xi X X, Kwok L Y, Sun Z H, Zhang H P, Zhang W Y. 2015. Evaluation of bacterial contamination in raw milk, ultra-high temperature milk and infant formula using single molecule, real-time sequencing technology. Journal of Dairy Science, 98(12): 8 464-8 472.
DOI:10.3168/jds.2015-9886 |
Howarth R W. 1993. Microbial processes in salt-marsh sediments. In: Ford T E ed. Aquatic Microbiology: an Ecological Approach. Blackwell Scientific Publications, Boston. p.239-260.
|
Itoh T, Suzuki K I, Nakase T. 1998. Thermocladium modestius gen. nov., sp. nov., a new genus of rod-shaped, extremely thermophilic crenarchaeote. International Journal of Systematic Bacteriology, 48(3): 879-887.
DOI:10.1099/00207713-48-3-879 |
Itoh T, Suzuki K I, Sanchez P C, Nakase T. 1999. Caldivirga maquilingensis gen. nov., sp. nov., a new genus of rodshaped crenarchaeote isolated from a hot spring in the Philippines. International Journal of Systematic Bacteriology, 49(3): 1 157-1 163.
DOI:10.1099/00207713-49-3-1157 |
Jørgensen B B. 1982. Mineralization of organic matter in the sea bed-the role of sulphate reduction. Nature, 296(5858): 643-645.
DOI:10.1038/296643a0 |
Kim M, Oh H S, Park S C, Chun J. 2014. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. International Journal of Systematic and Evolutionary Microbiology, 64: 346-351.
DOI:10.1099/ijs.0.059774-0 |
Klepac-Ceraj V, Bahr M, Crump B C, Teske A P, Hobbie J E, Polz M F. 2004. High overall diversity and dominance of microdiverse relationships in salt marsh sulphate-reducing bacteria. Environmental Microbiology, 6(7): 686-698.
DOI:10.1111/j.1462-2920.2004.00600.x |
Kotiaho M, Fritze H, Merilä P, Juottonen H, Leppälä M, Laine J, Laiho R, Yrjälä K, Tuittila E S. 2010. Methanogen activity in relation to water table level in two boreal fens. Biology and Fertility of Soils, 46(6): 567-575.
DOI:10.1007/s00374-010-0461-0 |
Langille M I G, Zaneveld J, Caporaso J G, McDonald D, Knights D, Reyes J A, Clemente J C, Burkepile D E, Vega T R L, Knight R, Beiko R G, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31(9): 814-824.
DOI:10.1038/nbt.2676 |
Larsen S, Nielsen L P, Schramm A. 2015. Cable bacteria associated with long-distance electron transport in New England salt marsh sediment. Environmental Microbiology, 7(2): 175-179.
DOI:10.1111/1758-2229.12216 |
Liu W J, Zheng Y, Kwok L Y, Sun Z H, Zhang J C, Guo Z, Hou Q C, Menhe B, Zhang H P. 2015. High-throughput sequencing for the detection of the bacterial and fungal diversity in Mongolian naturally fermented cow's milk in Russia. BMC Microbiology, 15: 45.
DOI:10.1186/s12866-015-0385-9 |
Louca S, Parfrey L W, Doebeli M. 2016. Decoupling function and taxonomy in the global ocean microbiome. Science, 353(6305): 1 272-1 277.
DOI:10.1126/science.aaf4507 |
Lücker S, Steger D, Kjeldsen K U, MacGregor B J, Wagner M, Loy A. 2007. Improved 16S rRNA-targeted probe set for analysis of sulfate-reducing bacteria by fluorescence in situ hybridization. Journal of Microbiological Methods, 69(3): 523-528.
DOI:10.1016/j.mimet.2007.02.009 |
Moreau J W, Zierenberg R A, Banfield J F. 2010. Diversity of dissimilatory sulfite reductase genes (dsrAB) in a salt marsh impacted by long-term acid mine drainage. Applied and Environmental Microbiology, 76(14): 4 819-4 828.
DOI:10.1128/AEM.03006-09 |
Mori K, Kim H, Kakegawa T, Hanada S. 2003. A novel lineage of sulfate-reducing microorganisms:Thermodesulfobiaceae fam. nov., Thermodesulfobium narugense, gen. nov., sp. nov., a new thermophilic isolate from a hot spring. Extremophiles, 7(4): 283-290.
DOI:10.1007/s00792-003-0320-0 |
Mosher J J, Bernberg E L, Shevchenko O, Kan J, Kaplan L A. 2013. Efficacy of a 3rd generation high-throughput sequencing platform for analyses of 16S rRNA genes from environmental samples. Journal of Microbiological Methods, 95(2): 175-181.
DOI:10.1016/j.mimet.2013.08.009 |
Muyzer G, Stams A J M. 2008. The ecology and biotechnology of sulphate-reducing bacteria. Nature Reviews Microbiology, 6(6): 441-454.
DOI:10.1038/nrmicro1892 |
Nielsen L B, Finster K, Welsh D T, Donelly A, Herbert R A, De Wit R, Lomstein B A A. 2001. Sulphate reduction and nitrogen fixation rates associated with roots, rhizomes and sediments from Zostera noltii and Spartina maritima meadows. Environmental Microbiology, 3(1): 63-71.
DOI:10.1046/j.1462-2920.2001.00160.x |
Nielsen L P, Risgaard-Petersen N, Fossing H, Christensen P B, Sayama M. 2010. Electric currents couple spatially separated biogeochemical processes in marine sediment. Nature, 463(7284): 1 071-1 074.
DOI:10.1038/nature08790 |
Ollivier B, Cayol J L, Fauque G. 2007. Sulphate-reducing bacteria from oil field environments and deep-sea hydrothermal vents. In: Barton L L, Hamilton W A eds.Sulphate-Reducing Bacteria: Environmental and Engineered Systems. Cambridge University Press, Cambridge. p.305-328.
|
Pester M, Knorr K H, Friedrich M W, Wagner M, Loy A. 2012. Sulfate-reducing microorganisms in wetlands-fameless actors in carbon cycling and climate change. Frontiers in Microbiology, 3: 72.
DOI:10.3389/fmicb.2012.00072 |
Poffenbarger H J, Needelman B A, Megonigal J P. 2011. Salinity influence on methane emissions from tidal marshes. Wetlands, 31(5): 831-842.
DOI:10.1007/s13157-011-0197-0 |
Rabus R, Hansen T A, Widdel F. 2006. Dissimilatory sulfateand sulfur-reducing prokaryotes. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K H, Stackebrandt E eds. The Prokaryotes, Vol. 2: Ecophysiology and Biochemistry.Springer, Berlin. p.659-768.
|
Rambaut A. 2016. FigTree v1.4.3 software. Institute of Evolutionary Biology, University of Edinburgh.Rooney-Varga J N, Devereux R, Evans R S, Hines M E. 1997.Seasonal changes in the relative abundance of uncultivated sulfate-reducing bacteria in a salt marsh sediment and in the rhizosphere of Spartina alterniflora. Applied and Environmental Microbiology, 63(10): 3 895-3 901.
|
Rooney-Varga J N, Devereux R, Evans R S, Hines M E. 1997. Seasonal changes in the relative abundance of uncultivated sulfate-reducing bacteria in a salt marsh sediment and in the rhizosphere of Spartina alterniflora. Applied and Environmental Microbiology, 63(10): 3 895-3 901.
|
Schauer R, Risgaard-Petersen N, Kjeldsen K U, Tataru Bjerg J J, Jørgensen B B, Schramm A, Nielsen L P. 2014. Succession of cable bacteria and electric currents in marine sediment. ISME Journal, 8(6): 1 314-1 322.
DOI:10.1038/ismej.2013.239 |
She C X, Zhang Z C, Cadillo-Quiroz H, Tong C. 2016. Factors regulating community composition of methanogens and sulfate-reducing bacteria in brackish marsh sediments in the Min River estuary, southeastern China. Estuarine, Coastal and Shelf Science, 181: 27-38.
DOI:10.1016/j.ecss.2016.08.003 |
Shen Y, Buick R. 2004. The antiquity of microbial sulfate reduction. Earth-Science Reviews, 64(3-4): 243-272.
DOI:10.1016/S0012-8252(03)00054-0 |
Silvestro D, Michalak I. 2012. RaxmlGUI:a graphical frontend for RAxML. Organisms Diversity & Evolution, 12(4): 335-337.
DOI:10.1007/s13127-011-0056-0 |
Smith J M, Green S J, Kelley C A, Prufert-Bebout L, Bebout B M. 2008. Shifts in methanogen community structure and function associated with long-term manipulation of sulfate and salinity in a hypersaline microbial mat. Environmental Microbiology, 10(2): 386-394.
DOI:10.1111/j.1462-2920.2007.01459.x |
Steudler P A, Peterson B J. 1984. Contribution of gaseous Sulphur from salt marshes to the global Sulphur cycle. Nature, 311(5985): 455-457.
DOI:10.1038/311455a0 |
Sulu-Gambari F, Seitaj D, Meysman F J R, Schauer R, Polerecky L, Slomp C P. 2016. Cable bacteria control ironphosphorus dynamics in sediments of a coastal hypoxic basin. Environmental Science & Technology, 50(3): 1 227-1 233.
DOI:10.1021/acs.est.5b04369 |
Wagner M, Amann R, Lemmer H, Schleifer K H. 1993. Probing activated sludge with oligonucleotides specific for proteobacteria:inadequacy of culture-dependent methods for describing microbial community structure. Applied and Environmental Microbiology, 59(5): 1 520-1 525.
DOI:10.1128/AEM.59.5.1520-1525.1993 |
Welsh D T, Bourguès S, De Wit R, Auby I. 1997. Effect of plant photosynthesis, carbon sources and ammonium availability on nitrogen fixation rates in the rhizosphere of Zostera noltii. Aquatic Microbial Ecology, 12(3): 285-290.
DOI:10.3354/ame012285 |
Yuan H W, Chen J F, Ye Y, Lou Z H, Jin A M, Chen X G, Jiang Z P, Lin Y S, Chen C T A, Loh P S. 2017. Sources and distribution of sedimentary organic matter along the Andong salt marsh, Hangzhou Bay. Journal Marine Systems, 174: 78-88.
DOI:10.1016/j.jmarsys.2017.06.001 |
Zeleke J, Sheng Q, Wang J G, Huang M Y, Xia F, Wu J H, Quan Z X. 2013. Effects of Spartina alterniflora invasion on the communities of methanogens and sulfate-reducing bacteria in estuarine marsh sediments. Frontiers in Microbiology, 4: 243.
DOI:10.3389/fmicb.2013.00243 |
Zhang X M, Zhou Y, Liu P, Wang F, Liu B J, Liu X J, Yang H S. 2015. Temporal pattern in biometrics and nutrient stoichiometry of the intertidal seagrass Zostera japonica and its adaptation to air exposure in a temperate marine lagoon (China):implications for restoration and management. Marine Pollution Bulletin, 94(1-2): 103-113.
DOI:10.1016/j.marpolbul.2015.03.004 |