 2021, Vol. 39
2021, Vol. 39Institute of Oceanology, Chinese Academy of Sciences
Article Information
- WANG Hongxia, YUE Xin, YU Jiajia, WANG Rui, TENG Shuangshuang, FANG Jun, LIU Baozhong
- Microbial community changes in the digestive tract of the clam Meretrix petechialis in response to Vibrio parahaemolyticus challenge
- Journal of Oceanology and Limnology, 39(1): 329-339
- http://dx.doi.org/10.1007/s00343-020-9217-3
Article History
- Received Aug. 29, 2019
- accepted in principle Jan. 15, 2020
- accepted for publication Apr. 22, 2020




2 Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266000, China;
3 Zhejiang Mariculture Research Institute, Wenzhou 325005, China;
4 Zhejiang Key Laboratory of Exploitation and Preservation of Coastal Bio-resource, Wenzhou 325005, China
According to the latest the State of World Fisheries and Aquaculture (SOFIA) report, bivalves are one of the most important food products for the aquaculture industry, and worldwide production (mainly oysters, mussels, clams, and scallops) reached a volume of 15 t with an economic value of more than $18 billion (FishStatJ, Food and Agriculture Organization of the United Nations (FAO) http://www.fao.org/fishery/statistics/global-aquaculture-production/en). China is one of the main bivalve-producing countries, and the clam culture industry has become very important for many regions in China. However, some infectious diseases have frequently occurred over the last decade and have become the primary limiting factors for the viability and development of the bivalve aquaculture industry. For example, Vibrio parahaemolyticus is an opportunistic pathogen that usually causes mass mortality of the cultured clam Meretrix petechialis in summer. This gram-negative bacterium isolated from diseased clams was demonstrated to be the main pathogenic by artificial infection, and a correlation exists between adult clam mortality and an increased concentration of V. parahaemolyticus (Yue et al., 2010).
As invertebrates, clams largely depend on innate immunity to defend against and resist infectious agents. The hepatopancreas is part of the digestive tract and an important member of the immune system in which epithelial cells produce the majority of immune molecules (Rőszer, 2014). In addition, epithelia lining the digestive tract of bivalves phagocytize biotic and abiotic particles and colloids, so it has dual function in nutrition and defense by promoting nutrient absorption and by keeping pathogens in check to improve disease resistance (Allam and Espinosa, 2016). Additionally, the digestive tract of bivalves has abundant microorganisms, including pathogens and commensals from the surrounding environment. For example, it was reported that oysters are capable of filtering over 10 L/(h∙g) dry tissue (Dupuy et al., 2000), which equated to over 25 000 microbes/s for a 1-g oyster in a modest 104 microbes/mL seawater (Allam and Espinosa, 2016). Therefore, the entire digestive tract microflora may have crucial effects on the health status of the host during vibrio infection.
It is becoming clear that diseases could substantially change the intestinal microbiota composition in both vertebrates and invertebrates (Yang et al., 2017; Xiong et al., 2018; Yu et al., 2019b). Studies on the microbiota-disease relationship in shrimp intestines revealed that dysbiosis in unhealthy intestinal microbiota preceded the appearance of physical symptoms of the disease, which emphasized that microorganisms are the first to respond to changes and can augment disease progression (Xiong, 2018). In addition, studies identified that the degree of change in intestinal bacterial communities of shrimp is closely associated with the disease severity (Xiong et al., 2015; Chen et al., 2017). Currently, research on bacterial community alterations in bivalves with vibrio disease is still rare and has mostly focused on hemolymph (Wendling et al., 2014; Lokmer and Wegner, 2015; Petton et al., 2015). Lokmer and Wegner (2015) previously reported that the microbial community of oyster hemolymph could be significantly affected by temperature stress, and more stable communities had greater resistance to invasion by pathogens. However, little is known about the role of the pathogen vibrio and the dynamics of how the microbial communities of the digestive tract change during disease progression when clams are infected with a high concentration of vibrio.
Increasing evidence has revealed complex interactions among pathogens, hosts and commensal microbiota (Boutin et al., 2013). The rapid development of high-throughput sequencing technologies and bioinformatic analyses facilitates indepth characterization of complex and varied microbiota (Caporaso et al., 2010; Boutin et al., 2013) and further development a broader understanding of the complexity of host-bacteria interactions. In this study, a 16S rRNA sequencing approach was used to investigate the dynamic changes of digestive tract bacterial communities in the clam M. petechialis subjected to bacterial challenge with V. parahaemolyticus. The microbiological data obtained in this study help us to understand the role and composition of host-associated microbial communities that are affected by vibrio infection, which is important for diagnosing, predicting, and preventing disease outbreaks in clam aquaculture.
2 MATERIAL AND METHOD 2.1 Experimental clamsIn total, 250 clams (M. petechialis) with a mean body weight of 30.76±8.01 g were used for the experiment. The clams were caught from the cultured population and brought into the laboratory. Before vibrio challenge, they were acclimated for 2 weeks in a holding system at 26 ℃ with salinity at 22.0±1.0 and continuously aerated seawater, and fed Isochrysis galbana. This holding system maintained water quality with a filtration unit.
2.2 Challenge experimentAt the beginning of the challenge experiment, the digestive tracts of 30 acclimated clams were arbitrarily sampled and used to assay the initial vibrio load. By colony-forming unit (CFU) counts, no vibrio load was detected in 83.3% of individuals (25/30), and a vibrio load between 0.08–1.26 CFU/mg tissue was detected in the other five clams (the detailed sampling and vibrio load assay methods are described in Section 2.3). Therefore, it could be deduced that there was seldom vibrio infection in the clams before the artificial infection. Vibrio challenge was conducted according to Liang et al. (2017). Briefly, approximately 150 clams were arbitrarily collected and divided into five groups. Each group of 28–32 clams was put in a single cage, and all clams from the five cages were reared in a water tank with V. parahaemolyticus strain MM21 (Yue et al., 2010) added to the tank at a final concentration of 1×107 CFU/mL at 30 ℃. Seawater mixed with vibrio was renewed once per day. Three groups were used to monitor daily mortality and the other two groups were used for sample collection. After 4 days (i.e., on day 5), the artificial vibrio challenge was stopped, and the clams were reared in fresh seawater again. Meanwhile, the remaining clams remained in a fresh seawater tank and were classified as the control group, which was used to confirm that mortality was due to pathogen exposure. Clams were checked every 6 h per day, dead or moribund individuals were collected and recorded. The daily mortality was calculated based on three replicates.
2.3 Sampling and vibrio load assayDuring the challenge experiment, three clams were collected from each of the two challenge groups and one control group at 1 day post infection (DPI), 3 DPI, 4 DPI, and 6 DPI respectively. All clams were checked for survival during sample collection. The digestive tract (whole hepatopancreas and stomach) of the clams was aseptically sampled, and each sample was dissected into two parts. One part was immediately frozen in liquid nitrogen and stored at -80 ℃ until it was used for 16S rRNA sequencing. The other part was immediately stored on ice, weighed, placed into 500 μL sterile seawater, and homogenized as soon as possible. The homogenates were diluted an appropriate number of times, 50 μL was then plated onto vibrio-selective thiosulfate-citrate-bile-saltssucrose (TCBS) agar, and the CFU were counted after 24 h of cultivation at 25 ℃.
2.4 PCR amplification and 16S rRNA sequencingMicrobial DNA of 15 samples (three samples per time point) was extracted using the CTAB/SDS method. Polymerase chain reaction (PCR) was performed with Phusion® High-Fidelity PCR Master Mix (New England Biolabs). The variable V4–V5 region of the bacterial 16S rRNA gene sequences were amplified with the primer pair 515F (5′GTGCCAGCMGCCGCGG3′) and 907R (5′CCGTCAATTCMTTTRAGTTT3′) with a barcode. All PCRs, purification, and library constructions were performed as previously described (Li et al., 2018). Finally, 250-bp paired-end reads were generated on the Illumina HiSeq2500 platform.
2.5 Pyrosequencing and data analysisThe raw sequence reads were filtered using the Quantitative Insights Into Microbial Ecology (QIIME) quality controlled process (Caporaso et al., 2010). Potential chimeric sequences were identified and removed by the UCHIME algorithm (Edgar et al., 2011). After filtering, the high-quality sequences obtained were used in the following bioinformatics analyses. The operational taxonomic units (OTUs) were clustered with a cutoff of 97% similarity using Uparse (Edgar, 2013). The representative (most abundant) sequence for each OTU was screened and then taxonomically classified in the SILVA database (http://www.arb-silva.de), using the Ribosomal Database Project classifier (at 80% confidence threshold) (Wang et al., 2007; Quast et al., 2013). Sequences classified to the genus Vibrio were then compared with 16S rRNA sequences of the cultured strain MM21 to identify that the OTU corresponded to the MM21 strain. Singleton OTUs and sequences identified as chloroplasts or mitochondria were removed from the analysis. All sequences obtained from this study were deposited in GenBank Sequence Read Archive (SRA; accession Nos. SRR8741669–SRR8741683).
2.6 Statistical analysisThe rarefaction curves and rank-abundance curves, which were plotted to determine the abundance of communities and sequencing data of each sample, were constructed using R (version 3.2.3, http://www.r-project.org). Alpha diversity (OTU richness, phylogenetic diversity, and Simpson index) and beta diversity were analyzed by QIIME with default parameters. Beta diversity across the 15 samples was calculated based on Bray-Curtis and weighted UniFrac distances (Hamady et al., 2010) and analyzed by nonmetric multidimensional scaling (NMDS), which showed the overall dissimilarity of the bacterial community in different infection stages. An unweighted pair group method with arithmetic mean (UPGMA) tree was also constructed with QIIME. Discriminatory analysis of taxonomic groups among infection stages was performed with LEfSe (P < 0.01, log 10 linear discriminant analysis score>4) (Segata et al., 2011).
3 RESULT 3.1 Mortality under vibrio challengeClams in the challenge group were infected with V. parahaemolyticus by immersion infection. Low mortality was observed (1.09%) during the first 2 days, but mortality then drastically increased and the daily mortality reached 16.3% on the fifth day. Although the artificial vibrio challenge was stopped on day 5, the daily mortality peaked at 34.78% and 29.35% on day 6 and 7, respectively (Fig. 1). Additionally, nearly 70% of surviving individuals on day 6 died by the following time point (day 7). Experimental challenge with V. parahaemolyticus resulted in 86.96% accumulated mortality of clams within 7 days, as opposed to no death events in the control group (P < 0.001). The results indicated that the controlled experiments of clam immersion in vibrio-contaminated seawater was an effective way to mimic the acute disease outbreak.

|
| Fig.1 Mortality curves for clams challenged with vibrio Letters above groups indicate significant differences in the means. |
We characterized the microbial dynamics and composition of the communities in the healthy clams (control group) and challenged clams at different time points. The key time points were 1, 3, and 4 DPI under constant vibrio immersion infection, as clams were healthy and only sporadically died in these stages; at 6 DPI, 1 day after stopping the artificial infection, a death outbreak occurred and the daily mortality peaked at this time point. Briefly, these time points covered different infection phases and were in consistent with those identified by a previous infection experiment (Yu et al., 2019a), these time points could be divided into latency phase (1 DPI, no dead clams found), prodrome phase (3 and 4 DPI, dead clams were sporadically observed), and onset phase (6 DPI, death outbreak occurred).
In total, 913 225 valid sequence reads binned into 1 733 OTUs were retrieved from 15 individual samples of the control and challenge groups over time. The rarefaction curves of most samples were nearly asymptotic, which indicated that the sequencing depth per sample covered most of the microorganisms (Fig. 2). A total of 120 OTUs were shared by all five groups, whereas there were 33 unique OTUs in the control group, 125 in the 1 DPI group, 485 in the 3 DPI group, 131 in the 4 DPI group, and 16 in the 6 DPI group (Fig. 3). Among the groups, the highest number of unique OTUs was observed in the 3 DPI group, which indicated that more specific microbial species appeared in this phase.

|
| Fig.2 Rarefaction curves of 15 samples The curves were generated based on a 97% sequence similarity threshold level of OTUs. |

|
| Fig.3 Numbers of shared and specific OTUs in the five groups that represent different infection stages |
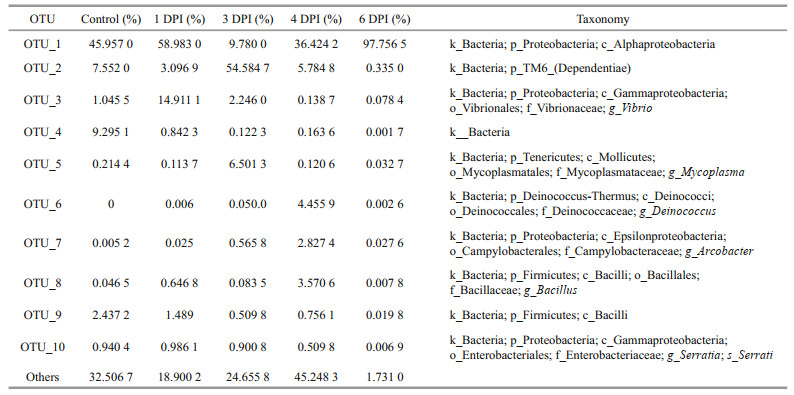
The relative OTU abundances fit the Fisher's logseries distribution well, which indicated that few OTUs accounted for the majority of the reads. For example, the relative abundance of OTU_1 (Alphaproteobacteria) was 49.78%±33.15% in the samples (Table 1). The phyla Proteobacteria and TM6-(Dependentiae) encompassed the bulk of the OTU diversity and abundance across all 15 samples from the five groups (Fig. 4). In detail, 67.5% of the phylotypes belonged to Proteobacteria, and 14.3% belonged to TM6-(Dependentiae). Proteobacteria and TM6-(Dependentiae), the two core phyla, were relatively similarly distributed in each sample in each group, except in one specimen, 4-1, which was vastly different from the other specimens in the 4 DPI group. The core phylum composition showed significant changes over the course of vibrio challenge. There was a significant decrease in Proteobacteria abundance, and TM6-(Dependentiae) abundance markedly increased and became the dominant bacteria at 3 DPI. In the subsequent stage, the Proteobacteria abundance increased again and accounted for 99% of the total community at 6 DPI. However, the composition of Proteobacteria at 6 DPI became far less diverse, with OTU_1 (Alphaproteobacteria) accounting for nearly 97.75%±0.25% of the bacterial diversity.
|

|
| Fig.4 Relative abundance of bacterial phyla in digestive tract samples at different stages of vibrio challenge |
During the course of vibrio challenge, a significant increase in the number of OTUs was observed at 3 DPI (P < 0.05), followed by a distinct decline at 6 DPI (P < 0.05). The phylogenetic diversity also significantly increased at 3 DPI compared with the control group (Fig. 5a), although the microbial communities of 1 and 4 DPI exhibited no significant variation compared with that of the healthy control group based on the alpha diversity analysis (Fig. 5a). The UPGMA tree based on weighted UniFrac distances, which measures the phylogenetic similarity between bacterial communities, showed that the microbiomes of the control group and 1 DPI group (initial time of challenge) clustered first, and the microbial communities in the 3 DPI group differed more from those of other groups (Fig. 5b). This finding was consistent with previous alpha diversity results (Fig. 5a). These results both demonstrated that a dramatic change in microbial communities in the digestive tract occurred at 3 DPI during vibrio challenge. Notably, a significantly low OTU diversity was found in the moribund samples at 6 DPI (Fig. 5a), which indicated that the communities of the moribund samples were characterized by the disappearance of some rare OTUs and proliferation of one OTU (OTU_1).

|
| Fig.5 Microbial diversity comparisons of digestive tract samples at different infection stages a. OTU richness, phylogenetic diversity and Simpson index; b. UPGMA clustering based on relative abundance at the phylum level. |
Furthermore, we connected the samples from one group and used the area of the triangles in the NMDS plot to represent the relative stability of community composition at different stages during vibrio challenge (Fig. 6). The microbiomes in the early stage of vibrio infection (e.g., 1 and 3 DPI), were more stable compared with the microbiomes at the end stage of vibrio infection. Moreover, the health status of the clams appeared divergence with the progression of the vibrio challenge. The daily mortality was very low during the first 3 days, but 17.44% of surviving samples at 4 DPI were dead by the next day. Therefore, the unstable microbial dynamics at 4 DPI and 6 DPI might be partially caused by the individual variations in health status at the end stage of vibrio infection.

|
| Fig.6 NMDS plots of Bray-Curtis distances between the microbial communities showing sample status stability Each triangle represents an infected stage, and each vertex represents one sample. |
In the samples from natural environments, vibrio was detected at a relative abundance of approximately 0.2%–1.8%. The microbiomes of samples (1 DPI, 3 DPI, and 4 DPI) immersed in vibrio-contaminated seawater were unexpectedly dominated by Alphaproteobacteria (which could not be further classified to the genus level) and phylum TM6-(Dependentiae), and not by vibrio. The highest relative abundance of vibrio (14.9%±8.8%) was found at 1 DPI (Table 1). However, the relative abundance of vibrio subsequently decreased to 2.2%±0.7% and 0.1%±0.06%, respectively, at 3 DPI and 4 DPI (Table 1), even if a high concentration of V. parahaemolyticus was maintained in the surrounding environment. The relatively high persistence of bacterial residents (e.g., Alphaproteobacteria) before and after the vibrio challenge might indicate that the microbiome was buffering the colonization and amplification of the pathogen vibrio. Furthermore, the absolute abundance of vibrio in the clam digestive tract (half of which were used for 16S rRNA gene amplicon pyrosequencing) was calculated by CFU counts (Fig. 7). The vibrio colony counts were closely correlated with the relative read abundances in the 16S rRNA gene amplicon pyrosequencing analysis (Spearman's ρ=0.65, P < 0.01).

|
| Fig.7 Vibrio CFU counts on TCBS agar at different infection stages |
We next asked whether any of these bacterial taxa were strongly associated with the disease progression. LEfSe (P < 0.01, log 10 linear discriminant analysis score>4) analysis was used to determine the indicator species (Fig. 8). These analyses identified 33 discriminatory taxa, of which the majority (21) distinguished the 4 DPI group from the other groups. The discriminating taxon for the control group was Actinobacteria, despite this phylum appearing in the entire dataset. The genus vibrio could be considered an indicator at 1 DPI, which indicated that vibrio abundances were associated with infection but not directly with mortality or disease. The 3 DPI group was largely distinguished by the presence of the phylum TM6-(Dependentiae) and the genus Mycoplasma, whereas the 4 DPI group was distinguished by high abundances of the families Comamonadaceae and Planococcaceae, order Sphingobacteriales, and phylum Planctomycetes. Together, these indicator taxa might have the potential to distinguish health status among samples during vibrio infection.

|
| Fig.8 Identified bacterial taxonomic biomarkers by LEfSe (P < 0.01, log10 linear discriminant analysis score>4) analysis |
Vibrio is a gram-negative bacterium that is widely distributed in marine environments and is recognized as one of the main opportunistic pathogens that causes mass mortality in cultured clams in summer (Yue et al., 2010). The population structure of family Vibrionaceae ('vibrios') among healthy marine invertebrates in coastal habitats has been explored, and the low specificity of animal-associated vibrio populations was detected by comparison of the vibrio populations collected from habitat water samples and animal samples (Preheim et al., 2011). This indicates that vibrios do not have a high degree of host preference, which supports the hypothesis that environmental dynamics are important factors in vibrio colonization. Therefore, we used vibrio immersion infection to mimic the vibrio outbreak in wild environment.
In this process, clams could easily filter exogenous bacteria from seawater, and mucus- and gill-associated microbiota may constitute a first line of defense against pathogens compared with infection via injection (Espinosa et al., 2016; Lokmer et al., 2016). Immersion infection by vibrio-contaminated seawater clearly caused approximately 87% mortality within 7 days. Faster and higher mortality occurred here compared with previous experimental infection (Liang et al., 2017), mainly because of higher seawater temperature in this study. Thus, laboratory immersion infection is helpful for investigating microbial diversity and dynamics during disease progression in the clam Meretrix petechialis.
Given the functional importance of clam digestive tract in nutrition digestion and innate immunity, it is necessary to investigate how the invading pathogenic vibrio changes the microbial communities of the digestive tract during disease progression. Therefore, in this study, we investigated the bacteria changes in the clam digestive tract when challenged with vibrio using a 16S rRNA sequencing approach. The phylum analysis revealed that Proteobacteria, TM6-(Dependentiae), Firmicutes Bacteroidetes, Actinobacteria, Tenericutes and Deinococcus-Thermus were the most common phyla in the healthy clam samples. Proteobacteria was the most abundant phylum and accounted for 58.57%±9.16% of the average relative abundance in the control group. The result was generally consistent with those of recent studies in which the resident microbiota of oyster species was characterized by 16S rRNA pyrosequencing (Fernández et al., 2014; Bernal et al., 2017).
In addition, our results revealed dramatic changes in the microbial communities during vibrio disease progression that preceded the mortality dynamics. At the end stage of vibrio infection, high mortality was accompanied by an obvious decrease in the microbial diversity of the digestive tract, and the overproliferation of OTU_1 (Alphaproteobacteria) indicated the collapse of digestive tract microbe homeostasis at 6 DPI. The decrease in the diversity of the digestive tract microbial community coincided with impaired health, and therefore could be as an indicator of health decline. This phenomenon has been found in various animals (Garnier et al., 2007; Chang et al., 2008; Green and Barnes, 2010). Lokmer and Wegner (2015) also found that dead or moribund oysters displayed signs of hemolymph microbiome structure disruption that was characterized by very low diversity and proliferation of a few taxa. Overall, our results indicate that the digestive tract microbial community is not merely the result of a filter-feeding lifestyle, that is, the microbiome is not simply determined by its surrounding environment. The stability of these digestive tract microfloras and their possible role in stimulating immunity or in competitive exclusion of external pathogens may have crucially affected the health status of clams during vibrio infection.
Host resistance refers to the ability to reduce pathogen replication and limit parasite growth through behavioral, morphological, and/or immunological mechanisms (Råberg et al., 2007). Therefore, the absolute number of pathogens (pathogen burden) is an important indicator for evaluating host resistance. Our study showed an obvious decrease in vibrio CFU numbers and relative vibrio abundance in the digestive tract at 3 DPI, which demonstrated that the digestive tract might take certain defense strategies to eliminate infectious agents (vibrio). Previous studies showed that oxidative burst and antimicrobial peptides release would be potent reactions for intestine immunity to eliminate pathogens in invertebrates (Ha et al., 2005; Schmitt et al., 2012; Yang et al., 2016). This could partially explained why vibrio pathogens do not undergo mass proliferation in later infection stages. However, further studies are needed to determine how clams discriminate between commensal and pathogenic bacteria during immune elimination. Furthermore, we found that the digestive tract microflora at 3 DPI showed larger adjustments in microbial composition following vibrio challenge. This shift in the composition of endogenous OTUs might help repress the proliferation of external pathogens (e.g., vibrio), on the other hand this shift might indicate that the previous homeostasis of microflora was destroyed. So, although the absolute and relative vibrio abundance decreased at 3 DPI and the following days, both the damage of activated immune defense imposed on host tissues and the disruption of microbial homeostasis still possibly resulted in massive mortalities happened at 5 DPI.
Dramatic changes took place in the digestive tract microflora during the transition from healthy to diseased clams, and one of the most important issues is whether sensitive assemblages could serve as indicators of host health. In this study, we found that more than 30 bacterial taxa had significantly different abundances between the control and challenged sample clams. ε-Proteobacteria are usually rare in coastal seawater and sediments (Campbell et al., 2011; Gobet et al., 2012), and they are rare in oyster stomach, gut and gill microbiota (King et al., 2012; Wegner et al., 2013; Fernández et al., 2014). Arcobacter spp. was also rare in our control samples. Therefore, the sharp increase in Arcobacter spp. abundance observed after 3 and 4 days of vibrio infection indicates an indirect but pronounced role in mortality. High abundance of Arcobacter spp. strains have also been reported in moribund oysters (Lokmer and Wegner, 2015), necrotic sponges (Fan et al., 2013), and starved abalones (Tanaka et al., 2004), which indicates that Arcobacter spp. can act as opportunistic pathogens when they occur in high enough densities (Olson et al., 2014). Similarly, Mycoplasma is usually considered pathogens, and was common but in low abundance in our control samples; Mycoplasma significantly increased with vibrio infection at 3 DPI. Therefore, it is possible that these opportunistic pathogens are commonly present in the digestive tract of healthy clams. The immune system (or physical barriers in the body) is mostly capable of fending them off when the dose is small, but opportunistic pathogens may become infectious and fatal when the hosts are immunosuppressed because of (a) biotic stresses (de Lorgeril et al., 2018).
5 CONCLUSIONTo date, little is known about the bacterial community in the clam. Our results provided insight into the bacterial community composition changes when the clams were infected with a high concentration of a vibrio strain and revealed potential taxa that indicate the disease progression. Our findings will provide useful information for diagnosing, predicting, and preventing disease outbreaks in clam aquaculture.
6 DATA AVAILABILITY STATEMENTRNA-seq data and amplicon sequences for microbiota analysis have been made available through the SRA database (accession Nos. SRR8741669–SRR8741683). Other data analyzed during this study are included in the supplementary file. Complementary information is available from the corresponding authors on reasonable request.
Allam B, Espinosa E P. 2016. Bivalve immunity and response to infections:are we looking at the right place?. Fish & Shellfish Immunology, 53: 4-12.
|
Bernal M G, Fernández N T, Lastra P E S, Marrero R M, Mazón-Suástegui J M. 2017. Streptomyces effect on the bacterial microbiota associated to Crassostrea sikamea oyster. Journal of Applied Microbiology, 122(3): 601-614.
DOI:10.1111/jam.13382 |
Boutin S, Bernatchez L, Audet C, Derôme D. 2013. Network analysis highlights complex interactions between pathogen, host and commensal microbiota. PLoS One, 8(12): e84772.
DOI:10.1371/journal.pone.0084772 |
Campbell B J, Yu L Y, Heidelberg J F, Kirchman D L. 2011. Activity of abundant and rare bacteria in a coastal ocean. Proceedings of the National Academy of Sciences of the United States of America, 108(31): 12776-12781.
DOI:10.1073/pnas.1101405108 |
Caporaso J G, Kuczynski J, Stombaugh J, Bittinger K, Bushman F D, Costello E K, Fierer N, Peña A G, Goodrich J K, Gordon J I, Huttley G A, Kelley S T, Knights D, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QⅡME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5): 335-336.
DOI:10.1038/nmeth.f.303 |
Chang J Y, Antonopoulos D A, Kalra A, Tonelli A, Khalife W, Schmidt T, Young V B. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. Journal of Infectious Diseases, 197(3): 435-438.
DOI:10.1086/525047 |
Chen W Y, Ng T H, Wu J H, Chen J W, Wang H C. 2017. Microbiome dynamics in a shrimp grow-out pond with possible outbreak of acute hepatopancreatic necrosis disease. Scientific Reports, 7: 9395.
DOI:10.1038/s41598-017-09923-6 |
de Lorgeril J, Lucasson A, Petton B, Toulza E, Montagnani C, Clerissi C, Vidal-Dupiol J, Chaparro C, Galinier R, Escoubas J M, Haffner P, Dégremont L, Charrière G M, Lafont M, Delort A, Vergnes A, Chiarello M, Faury N, Rubio T, Leroy M A, Pérignon A, Régler D, Morga B, Alunno-Bruscia M, Boudry P, Le Roux F, DestoumieuxGarzón D, Gueguen Y, Mitta G. 2018. Immunesuppression by OsHV-1 viral infection causes fatal bacteraemia in Pacific oysters. Nature Communications, 9: 4215.
DOI:10.1038/s41467-018-06659-3 |
Dupuy C, Vaquer A, Lam-Höai T, Rougier C, Mazouni N, Lautier J, Collos Y, Le Gall S. 2000. Feeding rate of the oyster Crassostrea gigas in a natural planktonic community of the Mediterranean Thau Lagoon. Marine Ecology Progress Series, 205: 171-184.
DOI:10.3354/meps205171 |
Edgar R C, Haas B J, Clemente J C, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16): 2194-2200.
DOI:10.1093/bioinformatics/btr381 |
Edgar R C. 2013. UPARSE:highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10): 996-998.
DOI:10.1038/nmeth.2604 |
Espinosa E P, Koller A, Allam B. 2016. Proteomic characterization of mucosal secretions in the eastern oyster, Crassostrea virginica. Journal of Proteomics, 132: 63-76.
DOI:10.1016/j.jprot.2015.11.018 |
Fan L, Liu M, Simister R, Webster N S, Thomas T. 2013. Marine microbial symbiosis heats up:the phylogenetic and functional response of a sponge holobiont to thermal stress. The ISME Journal, 7(5): 991-1002.
DOI:10.1038/ismej.2012.165 |
Fernández N T, Mazón-Suástegui J M, Vázquez-Juárez R, Ascencio-Valle F, Romero J. 2014. Changes in the composition and diversity of the bacterial microbiota associated with oysters (Crassostrea corteziensis, Crassostrea gigas and Crassostrea sikamea) during commercial production. FEMS Microbiology Ecology, 88(1): 69-83.
DOI:10.1111/1574-6941.12270 |
Garnier M, Labreuche Y, Garcia C, Robert M, Nicolas J L. 2007. Evidence for the involvement of pathogenic bacteria in summer mortalities of the Pacific oyster Crassostrea gigas. Microbial Ecology, 53(2): 187-196.
DOI:10.1007/s00248-006-9061-9 |
Gobet A, Böer S I, Huse S M, van Beusekom J E E, Quince C, Sogin M L, Boetius A, Ramette A. 2012. Diversity and dynamics of rare and of resident bacterial populations in coastal sands. The ISME Journal, 6(3): 542-553.
DOI:10.1038/ismej.2011.132 |
Green T J, Barnes A C. 2010. Bacterial diversity of the digestive gland of Sydney rock oysters, Saccostrea glomerata infected with the paramyxean parasite, Marteilia sydneyi. Journal of Applied Microbiology, 109(2): 613-622.
|
Ha E M, Oh C T, Bae Y S, Lee W J. 2005. A direct role for dual oxidase in Drosophila gut immunity. Science, 310(5749): 847-850.
DOI:10.1126/science.1117311 |
Hamady M, Lozupone C, Knight R. 2010. Fast UniFrac:facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. The ISME Journal, 4(1): 17-27.
DOI:10.1038/ismej.2009.97 |
King G M, Judd C, Kuske C R, Smith C. 2012. Analysis of stomach and gut microbiomes of the eastern oyster(Crassostrea virginica) from coastal Louisiana, USA. PLoS One, 7(12): e51475.
DOI:10.1371/journal.pone.0051475 |
Li H Y, Wang H, Wang H T, Xin P Y, Xu X H, Ma Y, Liu W P, Teng C Y, Jiang C L, Lou L P, Arnold W, Cralle L, Zhu Y G, Chu J F, Gilbert J A, Zhang Z J. 2018. The chemodiversity of paddy soil dissolved organic matter correlates with microbial community at continental scales. Microbiome, 6: 187.
DOI:10.1186/s40168-018-0561-x |
Liang B B, Jiang F J, Zhang S J, Yue X, Wang H X, Liu B Z. 2017. Genetic variation in vibrio resistance in the clam Meretrix petechialis under the challenge of Vibrio parahaemolyticus. Aquaculture, 468: 458-463.
DOI:10.1016/j.aquaculture.2016.10.037 |
Lokmer A, Kuenzel S, Baines J F, Wegner K M. 2016. The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environmental Microbiology, 18(3): 970-987.
DOI:10.1111/1462-2920.13163 |
Lokmer A, Wegner K M. 2015. Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. The ISME Journal, 9(3): 670-682.
DOI:10.1038/ismej.2014.160 |
Olson J B, Thacker R W, Gochfeld D J. 2014. Molecular community profiling reveals impacts of time, space, and disease status on the bacterial community associated with the Caribbean sponge Aplysina cauliformis. FEMS Microbiology Ecology, 87(1): 268-279.
DOI:10.1111/1574-6941.12222 |
Petton B, Bruto M, James A, Labreuche Y, Alunno-Bruscia M, Le Roux F. 2015. Crassostrea gigas mortality in France:the usual suspect, a herpes virus, may not be the killer in this polymicrobial opportunistic disease. Frontiers in Microbiology, 6: 686.
|
Preheim S P, Boucher Y, Wildschutte H, David L A, Veneziano D, Alm E J, Polz M F. 2011. Metapopulation structure of Vibrionaceae among coastal marine invertebrates. Environmental Microbiology, 13(1): 265-275.
DOI:10.1111/j.1462-2920.2010.02328.x |
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner F O. 2013. The SILVA ribosomal RNA gene database project:improved data processing and web-based tools. Nucleic Acids Research, 41(D1): D590-D596.
|
Råberg L, Sim D, Read A F. 2007. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science, 318(5851): 812-814.
DOI:10.1126/science.1148526 |
Rőszer T. 2014. The invertebrate midintestinal gland("hepatopancreas") is an evolutionary forerunner in the integration of immunity and metabolism. Cell and Tissue Research, 358(3): 685-695.
DOI:10.1007/s00441-014-1985-7 |
Schmitt P, Rosa R D, Duperthuy M, de Lorgeril J, Bachère E, Destoumieux-Garzón D. 2012. The antimicrobial defense of the Pacific oyster, Crassostrea gigas. How diversity may compensate for scarcity in the regulation of resident/pathogenic microflora. Frontiers in Microbiology, 3: 160.
|
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett W S, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biology, 12: R60.
DOI:10.1186/gb-2011-12-6-r60 |
Tanaka R, Ootsubo M, Sawabe T, Ezura Y, Tajima K. 2004. Biodiversity and in situ abundance of gut microflora of abalone (Haliotis discus hannai) determined by cultureindependent techniques. Aquaculture, 241(1-4): 453-463.
DOI:10.1016/j.aquaculture.2004.08.032 |
Wang Q, Garrity G M, Tiedje J M, Cole J R. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16): 5261-5267.
DOI:10.1128/AEM.00062-07 |
Wegner K M, Volkenborn N, Peter H, Eiler A. 2013. Disturbance induced decoupling between host genetics and composition of the associated microbiome. BMC Microbiology, 13: 252.
DOI:10.1186/1471-2180-13-252 |
Wendling C C, Batista F M, Wegner K M. 2014. Persistence, seasonal dynamics and pathogenic potential of vibrio communities from Pacific oyster hemolymph. PLoS One, 9(4): e94256.
DOI:10.1371/journal.pone.0094256 |
Xiong J B, Wang K, Wu J F, Qiuqian L L, Yang K J, Qian Y X, Zhang D M. 2015. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Applied Microbiology and Biotechnology, 99(16): 6911-6919.
DOI:10.1007/s00253-015-6632-z |
Xiong J B, Yu W N, Dai W F, Zhang J J, Qiu Q F, Ou C R. 2018. Quantitative prediction of shrimp disease incidence via the profiles of gut eukaryotic microbiota. Applied Microbiology and Biotechnology, 102(7): 3315-3326.
DOI:10.1007/s00253-018-8874-z |
Xiong J B. 2018. Progress in the gut microbiota in exploring shrimp disease pathogenesis and incidence. Applied Microbiology and Biotechnology, 102(17): 7343-7350.
DOI:10.1007/s00253-018-9199-7 |
Yang H T, Yang M C, Sun J J, Shi X Z, Zhao X F, Wang J X. 2016. Dual oxidases participate in the regulation of intestinal microbiotic homeostasis in the kuruma shrimp Marsupenaeus japonicus. Developmental & Comparative Immunology, 59: 153-163.
|
Yang H T, Zou S S, Zhai L J, Wang Y, Zhang F M, An L G, Yang G W. 2017. Pathogen invasion changes the intestinal microbiota composition and induces innate immune responses in the zebrafish intestine. Fish & Shellfish Immunology, 71: 35-42.
|
Yu J J, Wang H X, Yue X, Liu B Z. 2019a. Dynamic immune and metabolism response of clam Meretrix petechialis to Vibrio challenge revealed by a time series of transcriptome analysis. Fish & Shellfish Immunology, 94: 17-26.
|
Yu Z C, Liu C, Fu Q, Lu G X, Han S, Wang L L, Song L S. 2019b. The differences of bacterial communities in the tissues between healthy and diseased Yesso scallop(Patinopecten yessoensis). AMB Express, 9: 148.
DOI:10.1186/s13568-019-0870-x |
Yue X, Liu B Z, Xiang J H, Jia J T. 2010. Identification and characterization of the pathogenic effect of a Vibrio parahaemolyticus-related bacterium isolated from clam Meretrix meretrix with mass mortality. Journal of Invertebrate Pathology, 103(2): 109-115.
DOI:10.1016/j.jip.2009.11.008 |